プラグイン
プラグイン方式だから、安価で柔軟な運用を実現。
必要な人に、必要な時だけ、賢く利用。
しかも、ライセンスが切れてもデータは無くなりません。
便利な解析ツールをプラグインとして提供しており、必要な期間だけ購入して使うことができます。 GeneSpringなど他の商用解析ソフトと違って、ライセンスが切れた後でも、解析結果を無料のSubio Platformで見ることができます。
また、研究チーム内でデータ共有する場合も同様です。 ほとんどの人は無料のSubio Platformだけで、データ解析の議論と作業に参加できます。 オミクスデータの解析は人任せにせず、チーム全体で取り組むことで、ディスカッションを活発にし、発見のチャンスを広げ、チームの基礎体力が向上します。
Basic Plug-inの操作例
Basic Plug-inを使うと、フィルタリング、PCA、階層的クラスタリング、ヒートマップ、発現差解析、ベン図による比較などを、Subio Platform上で実行できます。
以下の動画では、RNA-Seqデータ解析チュートリアルと同じデータを使い、Basic Plug-inで可視化と比較解析を進める流れを紹介しています。 5日間無料お試しの前に、実際の操作感をご確認ください。
Basic Plug-in クイックチュートリアルの詳しい説明を見る
解析フローの中でプラグインの使いどころを確認する
Basic Plug-in、Advanced Plug-in、System Plug-inは、解析の目的やステップによって使いどころが異なります。 各プラグインがどの解析ステップで使われるのかを確認したい場合は、オミクスデータ解析ガイドをご覧ください。
5日間お試し
プラグインは4種類あり、それぞれ異なるツールのパッケージとなっています。 この3種類すべてについて、5日間無料でお試しいただけます。 下のフォームよりお申し込みください。 メールでシリアルナンバーが届きますので、これを使ってプラグインをアクティベートしてください。
トライアルライセンスをご利用になれるのは一台のコンピューターにつき一回限りです。
Basic Plug-in 50,800円/年 8,500円/30日
Basic Plug-inは、遺伝子発現をはじめとするさまざまなオミクスデータ解析で、幅広く使われている基本的かつ汎用的な統計解析ツールのパッケージです。
機能一覧
| フィルターで、測定値の信頼できない遺伝子を除く。 |
詳細
<p>統計解析をする前に、解析にとってノイズとなる遺伝子を Filter ツールを使って除きましょう。具体的には、すべてのサンプルにおいてシグナルがノイズ領域から出ない遺伝子や、発現変動しない遺伝子を除きます。</p>
<p>このツールの使い方がよくわからないときは、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</p>
|
|---|---|
| 2群間で、統計学的有意に発現差のある遺伝子を抽出する。 |
詳細
<p>Compare to Control ツールと Compare 2 Groups ツールは、Basic Plug-inの中でも Filter ツールの次に使用頻度が高いツールでしょう。Fold差や統計検定によって発現差がある遺伝子(DEG)を抽出し、Volcano Plot を描画します。検定は、one-sample t-test、Student's t-test、Welch's t-test、Mann-Whitney u-test が実行可能です。また、p-value は BH法による多重補正を行った q-value に変換できます。</p>
<p>このツールの使い方がよくわからないときは、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</p>
|
| グループ間で、統計学的有意に発現差のある遺伝子を抽出する。 |
詳細
<p>2郡間で発現差がある遺伝子を知りたい場合は、T検定を使いますが、3つ以上のグループ間で発現差があるかどうかを知りたい場合はANOVA(分散分析)を使うように、と統計学の教科書では言われています。(現実の解析における対応は別として)</p>
<p>ANOVAを実行すると、p-value または BH FDRにより補正された q-value で遺伝子を絞り込むことができます。より高度な検定手法を用いた大場合は、Datasheet タブからProcessed Sginal のテーブルを出力して、これをRなどの統計解析ソフトに取り込んで実行してください。実行して得られた結果は、遺伝子IDとP値のテーブルを値付きの Measurement List としてインポートできます。</p>
<p>このツールの使い方がよくわからないときは、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</p>
|
| 2群間での発現差解析を、まとめて実行する。 |
詳細
<p>発現差のある遺伝子を、1対Nまたは総当たりの組み合わせでまとめて抽出することができます。大量の組み合わせがあるときにとても便利なツールです</p>
<p>このツールの使い方がよくわからないときは、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</p>
|
| 選択中の遺伝子から、挙動の似ている遺伝子を抽出する。 |
詳細
<p>Find Similar Patterns ツールは、選択している遺伝子(または遺伝子群の平均パターン)と、発現パターンがよく似ている/逆向きの遺伝子を検索・抽出するものです。近似度の測定方法として相関係数(Pearson Correlation)、コサイン類似度(Uncentered Correlation)、スピアマンの順位相関係数(Spearman Correlation)から選べます。</p>
<p>このツールの使い方がよくわからないときは、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</p>
<h3>新しいバージョンでの変更点</h3>
<p>積算曲線ではなく、ヒストグラムを描画するようになっています。</p>
|
| ツリークラスタリング:パターンの似た遺伝子グループを概観する。 |
詳細
<p>階層型クラスタリングは発現パターンの似ている遺伝子郡をグループにまとめたり、発現プロファイルの似ているサンプル群をグループにまとめたりして、全体像を大まかに把握するのに便利です。</p>
<p><strong>00:00</strong> Tree Clustering を実行する前に、下記のようなノイズとなる遺伝子群を除去する</p>
<ol><li>値が低すぎて、測定値が信用できない遺伝子</li>
<li>発現変動しない遺伝子</li>
</ol>
<p><strong>01:20</strong> QC2 のリストを使ってクラスタリングを実行する</p>
<p><strong>01:55</strong> サンプルは、発現プロファイルが normal と tumor の二つに大きく分かれることがわかる<br /><span class="Apple-tab-span" style="white-space:pre"> </span>ここで、normal で発現量が高く、tumor で低くなっている遺伝子を抽出する</p>
<p><strong>02:20</strong> 患者によって normal における発現量は異なるものの、tumor で減少するという向きは共通している<br /><span class="Apple-tab-span" style="white-space: pre;"> </span>特定の発現パターンを持つ遺伝子のグループを抽出した後、それらの「発現量」を見てみる</p>
<p><strong>02:50</strong> Cluster1 として抽出した遺伝子群に対して、再度クラスタリングを実行<br /><span class="Apple-tab-span" style="white-space: pre;"> </span>ただし、making ratio に属するノーマライズブロックを除去して、近似度の測定は"Euclidian"を用いる</p>
<p><span class="Apple-tab-span" style="white-space:pre"> </span>発現パターンが似ている遺伝子群が、今度は発現レベルによって細分化された</p>
<p><strong>03:55</strong> 別の角度からデータを見るたね、主成分分析(PCA)を実行する<br /><span class="Apple-tab-span" style="white-space:pre"> </span>第一主成分(PC1)はnormal と tumor の差を反映している。<br /><span class="Apple-tab-span" style="white-space:pre"> </span>一方第二主成分(PC2)は、tumorのサンプル群にサブグループが存在することを示唆する</p>
<p><strong>04:15</strong> 第二主成分(PC2)に貢献度の高い遺伝子を抽出する</p>
<p><strong>04:45</strong> PC2 contributing の遺伝子群を使ってクラスタリングを再度実行する</p>
<p><strong>05:00</strong> tumor のサブグループと、それらを分けている遺伝子の発現プロファイルが見える</p>
<p>このツールの使い方がよくわからないときは、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</p>
|
| 主成分分析(PCA):発現プロファイルの違いを概観する。 |
詳細
<p>主成分分析(PCA)は発現プロファイルによるサンプル間の近似度を視覚化します。近くにあるサンプル同士は発現プロファイルが似ています。また、0を境にスコアが正の方向と負の方向に分かれている場合は、逆向きに動いていることを表しています。</p>
<p>このツールの使い方がよくわからないときは、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</p>
|
Advanced Plug-in 50,800円/年 8,500円/30日
Advanced Plug-inは、Basic Plug-inによる解析結果を生物学的に解釈するのに便利なツールのパッケージです。 GO解析、パスウェイ解析、ゲノムの位置や配列に基づく解析などができます。
機能一覧
| 遺伝子リストから関連の示唆されるGOやパスウェイを検索する。 |
詳細
<p><strong>Enrichment Analysis</strong> tool は、抽出した遺伝子リストを生物学的コンテキストで解釈するのによく使われます。Gene Ontology (GO)はもちろんですが、パスウェイやサイトバンド、タンパクドメイン、miRNAのターゲットや転写因子のターゲット検索など、幅広い用途に使える便利なツールです。<a href="https://david-d.ncifcrf.gov/" title="https://david-d.ncifcrf.gov/" target="_blank">David Functional Annotation</a>は無料で使えるウェブツールで、私たちもこのツールをお勧めしていますが、Subio Platformのプラグインを使うとさらに便利な使い方ができます。</p>
<p>
なお、DAVID Functional Annotationは、この動画を作成した当時よりも使い勝手が大幅に向上しています。
現在のDAVIDを使った解析例については、
RNA-Seqチュートリアルの
<a href="/ja/info_technical/344#section7" title="RNA-Seqデータ解析チュートリアル">「7. 遺伝子アノテーションとエンリッチメント解析」</a>
もご覧ください。
</p>
<p>結果のテーブルには、Redanduncy90というスコアがあり、どの程度重複度の高いmeasurement listかを表しています。値は、他の検索対象のリストと重複するmeasurementの数の90th percentileです。比較的ユニークなリストが欲しい場合は、このスコアの低いmeasurement list を選択するといいでしょう。</p>
|
|---|---|
| パスウェイを取り込み、図中の遺伝子の発現パターンを重ねて表示する。 |
詳細
<p>Pathway Edit Tool は、任意の画像上にデータをヒートマップまたは棒グラフで重ねて表示する Pathway オブジェクトを作成・編集するものです。このツールは汎用的に作られているのでどのパスウェイにも対応できるのですが、まずは "KEGG Pathway Converter" ツールを別途提供して、KEGG パスウェイをより簡単にインポートできるようにしています。ご要望があれば、他のコンバーターツールも作成可能ですので、お気軽にご相談ください。</p>
<p>このツールの使い方がよくわからないときは、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</p>
<h3>新しいバージョンでの変更点</h3>
<p>KEGG Pathway Converterを別途ダウンロードする必要はありません。Subio Platformのインストールディレクトリに入っています。</p>
|
| がんステージ・年齢・用量などと、相関する遺伝子を抽出する。 |
詳細
<p><strong>Genes Tied in Parameter</strong> ツールは、たとえば年齢、処理後の経過時間、生存期間などの数値パラメータと、発現パターンが相関あるいは逆相関の関係になっている遺伝子を抽出します。</p>
<p><span style="background-color: transparent;">このツールの使い方がよくわからないときは、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</span>
</p>
|
| ゲノム上の相対的な位置関係から、遺伝子やプローブなどを抽出する。 |
詳細
<p>Geomic Location Filter ツールは、ゲノム上の相対的な位置に基づいて、ゲノム上のエレメント(遺伝子、プローブ、結合部位、メチル化サイトなど)を抽出するのに使います。このチュートリアルムービーでは、PHF8の結合部位を遺伝子の上流 2000 bp の範囲から抽出したり、逆にPHF8の結合部位を上流 2000 bp 以内に持つ遺伝子を抽出したりします。そして最後にその遺伝子の発現プローブを抽出して、これらの遺伝子について発現解析を行う準備をします。</p>
<p>このツールの使い方がよくわからないときは、無料<a href="/support" title="/support">オンラインサポート</a>をお申し込みください。</p>
|
| 染色体に沿って、値が高い・低い領域を視覚化する。 |
詳細
<p><strong>Summarize </strong>ツールを使うと、近接するタグやプローブをまとめて平均値などにして、全体的なトレンドを視覚化できます。たとえば、遺伝子発現やDNAメチル化の度合いが高く(または低く)なっている染色体領域を把握するのにとても便利です。</p>
<p>まとめる単位は、遺伝子の転写領域や、コーディング領域、上流域、下流域、あるいは決まったサイズのゲノムビンごとなど、汎用的に使えるようになっています。タイリングアレイ、CGHアレイ、ChIPやメチレーションなどさまざまなデータ解析で使えます。</p>
<p>もうひとつ「近傍にあるものをまとめる」ツールとして、<strong>Create Intervals </strong>ツールがあります。ゲノム上で近傍に密集しているタグやプローブは値が似ていることと考えられることがあります。このようなものをまとめることで Measurement の数を減らして解析しやすくすることができます。</p>
<p>これらのツールをうまく使うと、Measurement の数が多すぎてメモリーが足りなくて解析不能なデータが、解析可能になる場合があります。</p>
<p>このツールの使い方がよくわからない場合は、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</p>
|
| ゲノム上の複数の領域を指定して、そのゲノム配列をFASTAにする。 |
詳細
<p>特定の発現パターンを示す遺伝子リストを抽出した後、それら遺伝子群の上流域の塩基配列がほしい場合があります。Get Sequence ツールは、そのような遺伝子群を指定して相対的な位置の塩基配列を取得するものです。</p>
<p>このツールの使い方がよくわからないときは、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</p>
|
| ゲノム上で、指定した塩基配列の個所を検索する。 |
詳細
<p>Find Regions from Seq ツールは、特定の塩基配列を検索して、その位置を Region List として保存するものです。このムービーでは、FOXO1の制御配列を遺伝子の上流域に対して検索し、FOXO1によって制御されている可能性のある遺伝子を抽出します。</p>
<p>このツールの使い方がよくわからないときは、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</p>
|
| Genomeのデータから、measurementにアノテーションをつける。 |
詳細
<p>タイリングアレイ、ChIP-chip、CGH アレイ、メチレーションアレイ、ChIP-Seq、Methyl-Seqなどのデータを解析するとき、ただの位置情報だけあって遺伝子のアノテーションがないことがよくあります。</p>
<p>そのようなときは、Annotate Measurements ツールを使って近傍の遺伝子のアノテーションを転記すると、生物学的に解釈しやすくなります。</p>
<p>このツールの使い方がよくわからないときは、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</p>
|
| miRNAとターゲット遺伝子で、発現パターンが逆向きのペアを検索する。 |
詳細
<p><strong>Find miRNA Targets</strong> ツールは、遺伝子とmiRNAの実験データが対でそろっている場合に使えます。ここでは、実験的に検証されたmiRNAとターゲット遺伝子のペアや、計算によって予測されたmiRNAとターゲット遺伝子のペアのうち、発現パターンが逆相関になっているものを抽出します。すべてのペアについて相関係数の分布が出ますので、全体の傾向もつかむことができます。</p>
<p><a href="https://static.subioplatform.com/ssa/GSE34679.ssa" title="https://static.subioplatform.com/ssa/GSE34679.ssa">遺伝子発現データ(GSE34679)</a>と<a href="https://static.subioplatform.com/ssa/GSE34680.ssa" title="https://static.subioplatform.com/ssa/GSE34680.ssa">miRNAの発現データ(GSE34680)</a>のSSAファイルをダウンロードできるので、あなたのコンピューターで試してみてください。</p>
<p>このツールの使い方がよくわからないときは、無料<a href="/support" title="/support">オンラインサポート</a>をお申し込みください。</p>
|
| 遺伝子と近傍の因子で、互いのパターンが相関するペアを探す。 |
詳細
<p>Fnd Correlated Regions ツールは、たとえば ChIP-Seq(またはChIP-chip)と遺伝子発現のデータが対でそろっているときや、DNAメチル化データと遺伝子発現データが対でそろっているときなどに、発現パターンを制御している可能性の高い制御因子を抽出することができます。具体的には、遺伝子の発現パターンとその上流や重複する領域の制御因子のパターンの間の相関を計算し、相関関係あるいは逆相関関係にあるペアを抽出します。すべてのペアの相関係数の分布が表示されるので、全体の傾向を把握するのにも有用です。</p>
<p>このツールの使い方がよくわからないときは、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</p>
|
| 異なる種類のオミクスデータを統合して、散布図を描く。 |
詳細
<p>異なるタイプのオミクスデータでも Region List に変換しておけば、Scatter Plot of Regions ツールを使って直接比較することができます。</p>
<p>たとえば、遺伝子発現パターンと、その上流域のメチル化サイトのパターンや、染色体のコピー数とその領域にある遺伝子の発現パターンの関係を全体的に把握することができ、その影響が一部の遺伝子に限ったものなのか、一般化される支配的なものなのかを知ることができます。</p>
<p>このツールの使い方がよくわからないときは、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</p>
|
| カプランマイヤー生存曲線を描画する。 |
詳細
<p>Kaplan-Meier Survival Curve Tool を使って、カプランマイヤー生存曲線を描画するための準備について解説します。</p>
<p>ある治療を実施したグループとそうでないグループに分けて生存率を比較するなどはこれまで沢山の報告があります。Subio Platformに蓄積されたオミクスデータを使えば、たとえば、特定の遺伝子が高発現しているグループとそうでないグループや、がん抑制遺伝子のメチル化状態によって分けたグループで生存率を比較するなどがとても簡単にできます。</p>
<p>このツールの使い方がよくわからないときは、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</p>
|
System Plug-in 50,800円/年 8,500円/30日
System Plug-inは、前向き研究を実施している方や、大量のデータを蓄積してリファレンスとして利用している方、マルチオミクスデータの統合解析をされる方にとって必要な機能拡張です。
機能一覧
| MSigDBをインポートする / キーワードでMeasurement ListやPathwayを探す。 |
詳細
<p><span style="background-color: transparent;">Molecular Signatures Database (MSigDB) は、GSEAソフトウェアで使用するアノテーションされた遺伝子セットのコレクションで、大きく8つに分類されます。</span>
</p>
<ul><li>Hall mark<span style="background-color: transparent;"> gene sets, よく知られた生物学的状態あるいはプロセス</span>
</li>
<li>Positional gene sets, 染色体やサイトバンド</li>
<li>Curated gene sets, 公共のパスウェイデータ、論文、ドメインなど</li>
<li>Motif gene sets, 転写因子やmiRNAのシスエレメント</li>
<li>Computational gene sets, 癌由来の大量のマイクロアレイデータの解析で定義されたもの</li>
<li>GO gene sets, Gene Ontologyの遺伝子アノテーション</li>
<li>Oncogenic gene sets, 癌遺伝子を発現を操作したマイクロアレイ実験の結果で定義されたもの</li>
<li>Immunologic gene sets, 免疫研究のマイクロアレイによる発現データの解析で定義されたもの</li>
</ul>
<p>詳しくはこちらをご覧ください。<a href="https://software.broadinstitute.org/gsea/msigdb" title="https://software.broadinstitute.org/gsea/msigdb">https://software.broadinstitute.org/gsea/msigdb</a>
</p>
<p>上記の遺伝子セットをMeasurement List として取り込むことで、より強力なエンリッチメント解析が実行できるようになります。</p>
<p>"Find Measurement Lists or Pathways tool" は、その名前や含まれる遺伝子名からMeasurement ListやPathwayを検索することができる便利なツールです。</p>
<p>結果のテーブルには、Redanduncy90というスコアがあり、どの程度重複度の高いmeasurement listかを表しています。値は、他の検索対象のリストと重複するmeasurementの数の90th percentileです。比較的ユニークなリストが欲しい場合は、このスコアの低いmeasurement list を選択するといいでしょう。</p>
|
|---|---|
| Subio Platformに蓄積された大量のマルチオミクスデータを俯瞰する。 |
詳細
<p>大量のSeriesが、複数のPlatformやOrganismにまたがって存在する場合、一つ一つのSeriesを開いて見ていくのは非常に困難になります。</p>
<p><strong>Scan genes over series</strong> ツールは、そのような時に鳥観図のような視座を提供してくれます。簡単に言えば、<a href="https://www.ncbi.nlm.nih.gov/geoprofiles/" title="https://www.ncbi.nlm.nih.gov/geoprofiles/" target="_blank">GEO Profilesツール</a>と同じようなことが、自分のSubio Platformに蓄積しているデータに対して行えることです。違いは、一つの遺伝子だけでなく、二つの遺伝子名を同時に検索して比較できることです。このツールを使うと、下記のような質問に答えることができます。</p>
<ul><li>その遺伝子の発現が非常に高いSeriesはどれか?</li>
<li>その遺伝子が大きく変動するSeriesはどれか?</li>
<li>それら二つの遺伝子の発現パターンがよく似ているSeriesはどれか?</li>
</ul>
<p>
よく調べてみたいSeriesが見つかったら、Loadボタンを押して詳しく見てください。または、そのSeriesをSSAファイルに出力できます。</p>
<h3>このツールをうまく使うための注意点</h3>
<ul><li><strong>グラフの縦軸はCh1 Raw Signalの値です。</strong>したがって、GeneChipなら<a href="/ja/products/subioplatform/mas5-vs-rma" title="/ja/products/subioplatform/mas5-vs-rma">RMAよりMAS5</a>、RNA-Seqなら<a href="/ja/products/subioplatform/the-dynamic-range-of-rna-seq" title="/ja/products/subioplatform/the-dynamic-range-of-rna-seq">FPKM/TPMよりGene Count</a>というように、可能な限り<strong>生データに近い値</strong>をインポートして蓄積するようにして下さい。</li>
<li><strong>検索するのはGene Symbolです。</strong>Feature IDとして、マイクロアレイならProbe ID、RNA-SeqならENSEMBL Gene IDなどが使われていることが多いですので、PlatformリストのツールバーにあるSelect Symbol ColumnボタンでGene Symbolの格納されている列を選択してください。</li>
<li><strong>表示されるのは先頭に置いてあるDataSetです。</strong>そのため、すべてのSeriesにおいて、DataSetsセクションの先頭には"All Samples"という名前のDataSetを配置しておくことを推奨します。この"All Samples" DataSetを作成する際は、以下の設定を行ってください。</li>
</ul>
<ol><li><strong>個別サンプルの表示: </strong>サンプルグループによる平均化を行わず、個々のサンプルを表示させる設定にしてください。</li>
<li><strong>パラメーターの表示: </strong>そのSeriesの全体像を把握するために必要なパラメーターがすべて表示されるように設定してください。</li>
</ol>
<ul><li><strong>検索用データベースの作成に時間がかかります。</strong>このツールを使おうと思ってもすぐには使えません。帰る前や休み時間の前にデータベース構築を始めるなどして、使える状態にしてください。</li>
</ul>
<p>このツールの使い方がよくわからないときは、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</p>
|
| さまざまな統計値でTSS Plotを描画する。 |
詳細
<p>TSS Plot ツールは、何らかの値とTSSからの相対距離の関係を視覚化するものです。</p>
<p>よくあるのは、転写調節因子の結合や、DNAまたはヒストンの就職などのイベントの数をヒストグラムで表した図です。Subio PaltformのTSS Plotでは、さらに、メチル化比率や、メチル化状態の上昇または減退といった変化、遺伝子発現パターンとメチル化状態の変化パターンの相関係数など、より幅広い用途でお使いいただけます。</p>
|
| Seriesをコピーする。Seriesに含まれるSampleを追加/削除する。 |
詳細
<p>大量のSampleやSeriesがある場合に、これらのユーティリティツールは非常に便利です。</p>
<p><strong>Make A Copy of This Series</strong> は単純なツールです。しかし、Serieに変更を加える前にコピーしておくのは、安全に作業を進めるために必要です。</p>
<p><strong>Add/Remove Samples</strong> は、Seriesから質の悪いSampleを除去することができます。逆に、新しいSampleを追加することもできるので、これは前向き研究を実施している場合などには必須です。</p>
<p>このツールの使い方がよくわからないときは、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</p>
|
| 複数のPlatformを跨るマルチオミクスデータの統合解析の準備をする。 |
詳細
<p>このツールは、複数のオミックスデータセットを統合するために、さまざまなプラットフォームで共通のサンプルを見つけて印を付けます。このツールは<a href="/ja/info_technical/153" title="/ja/info_technical/153">Find miRNA Targets</a> や <a href="/ja/info_technical/159" title="/ja/info_technical/159">Find Correlated Regions</a> を使って解析を行うための準備を簡単に行えるようにするユーティリティです。</p>
|
| MeasurementをRegionに変換する。Regionにフィルターをかける。 |
詳細
<p>Seriesは一度にひとつずつしかロードできませんので、あるSeriesで使っていた Measurement List や DataSet は、別の Series をロードすると利用できなくなります。そこで、これらのデータを Region List に変換しておくと、Seriesをまたがってデータを比較することができるようになります。マルチオミクスデータの統合的な解析を可能にします。</p>
<p>また、Advanced Plug-in に含まれる多くのツールがインプットとして Region List を受け付けるようになっているので、そのようなツールを使う場合は、その前処理としてこのツールによるデータ変換が必要です。</p>
<p>このツールの使い方がよくわからないときは、無料<a href="/ja/support" title="/ja/support">オンラインサポート</a>をお申し込みください。</p>
<h3>新しいバージョンでの変更点</h3>
<p>Region Score Filterではフィルター対象の値をヒストグラムで表示し、閾値を適切に設定できるようになっています。</p>
|
| 外部のファイルから遺伝子リストをインポートする。 |
詳細
<p>テキストファイルから、一つまたは複数の遺伝子リストをSubio Platformにインポートして、これらの遺伝子群の発現パターンを概観することができます。また、遺伝子リストをインポートすると、これらを<a href="/ja/info_technical/152" title="/ja/info_technical/152">エンリッチメント解析</a>に利用できるようになります。</p>
<p>遺伝子のIDは、Platformに登録されている情報であればなんでもかまいません。また、遺伝子リストに何らかの統計値や数値が付加されている場合は、これらをフィルタリングに用いることができます。つまり、Rや他の統計ツールで計算した結果をSubio Platformに集合させることで、生物学的な解釈と強力な統計計算を統合することができるのです。</p>
|
Variation Plug-in 50,800円/年 8,500円/30日
Variation Plug-inは、FASTQファイルからSNPやindelを検出したり、変異のアノテーションを行ったり、変異を集計して候補を素早く抽出できるようにするためのツールのパッケージです。
Feature List
| RNA-SeqのFASTQファイルから、ゲノム変異(SNP や indel) を検出する。 |
詳細
<p>このパイプラインは、<a href="https://gatk.broadinstitute.org/hc/en-us/articles/360035531192-RNAseq-short-variant-discovery-SNPs-Indels-" title="https://gatk.broadinstitute.org/hc/en-us/articles/360035531192-RNAseq-short-variant-discovery-SNPs-Indels-" target="_blank">GATKのRNAseq short variant discovery (SNPs + Indels)</a>を参考にして作っています。ただし、メモリーの消費量を抑えるため、アラインメントにはSTARではなくHISAT2を使っています。ウェットのバイオロジストにとってGATKを動かすのは簡単ではありませんが、特にWindows ユーザーにとっては、このツールを使うのがもっとも簡単だと思います。ただし、ヒトのRNA-Seqデータにしか使えません。他の生物種ではご利用いただけませんのでご注意ください。</p>
<p>実行する前に、下記に従って実行環境のセットアップを行ってください。</p>
<ul><li><a href="/ja/info_technical/359" title="/ja/info_technical/359">Windows でのセットアップガイド</a>
</li>
<li><a href="/ja/info_technical/358" title="/ja/info_technical/358">macOS でのセットアップガイド</a>
</li>
</ul>
|
|---|---|
| 検出された SNP や indel がもたらす影響を予測する。 |
詳細
<p><a href="https://snpeff.sourceforge.net/" title="https://snpeff.sourceforge.net/" target="_blank">SnpEffは 変異に対してアノテーションとそれがもたらす効果を予測するツールです。</a> Annotate VCF ツールは、このプログラムを Subio Platform から簡単に実行できるようにするものです。ムービーではGZで圧縮されたファイルでもいいとなっていますが、解凍したVCFファイルでないとうまく動作しないかもしれません。</p>
<p>もし、解析対象の生物種用のデータベースが見つからない場合は、<a href="https://snpeff.sourceforge.net/SnpEff_manual.html#databases" title="https://snpeff.sourceforge.net/SnpEff_manual.html#databases" target="_blank">SnpEff用のデータベースの作成を行ってください</a>。</p>
|
| 変異をケースとコントロールで比較し、フィルタリングする。 |
詳細
<p>このツールは、注釈付きのVCFファイルを受け付けます。したがって、まず Annotate VCF ツールを実行してください。</p>
<p>インプットのVCFファイル群は、ケースグループとコントロールグループの2つのグループに分かれているかもしれません。このツールは、突然変異の種類と、コントロールグループとケースグループにおける出現頻度で集計し、フィルタリングをかけられるようにすることで、ターゲット候補の変異を簡単に見つけられるようにします。</p>
<p>また、2つの実行モードがあります。Count by Variant モードでは、変異ごとに集計します。一方、Count by Location モードでは、どのような変異かは無視して、位置だけを考慮して集計します。</p>
|
| 遺伝子ごとに変異をケースとコントロールで比較し、フィルタリングする。 |
詳細
<p>このツールは、注釈付きのVCFファイルを受け付けます。したがって、まず Annotate VCF ツールを実行してください。</p>
<p>インプットのVCFファイル群は、ケースグループとコントロールグループの2つのグループに分かれているかもしれません。このツールは、突然変異の種類と、コントロールグループとケースグループにおける出現頻度を遺伝子ごとに集計し、フィルタリングをかけられるようにすることで、ターゲット候補の遺伝子を簡単に見つけられるようにします。</p>
|
| ターゲット遺伝子のエクソンごとに変異を集計する。特定のエクソン上の変異を抽出する。 |
詳細
<p>このツールは、Aggregate Variants や Aggregate Variants per Gene ツールで解析した後に使用してください。<span style="background-color: transparent;">対象遺伝子の候補をリストアップした後、候補ごとにエクソンごとに要約することができます。</span>
</p>
<p>また、エクソンによるフィルタツールは、指定された転写物の指定されたエクソン上にあるゲノム要素を抽出します。</p>
|
価格
プラグイン
| 1年ライセンス | 30日ライセンス | |
|---|---|---|
| Basic Plug-in | 358 USD | 59 USD |
| Advanced Plug-in | 358 USD | 59 USD |
| System Plug-in | 358 USD | 59 USD |
| Variation Plug-in | 358 USD | 59 USD |
| 1年ライセンス | 30日ライセンス | |
|---|---|---|
| Basic Plug-in | 358 EUR | 59 EUR |
| Advanced Plug-in | 358 EUR | 59 EUR |
| System Plug-in | 358 EUR | 59 EUR |
| Variation Plug-in | 358 EUR | 59 EUR |
| 1年ライセンス | 30日ライセンス | |
|---|---|---|
| Basic Plug-in | 50,800 JPY | 8,500 JPY |
| Advanced Plug-in | 50,800 JPY | 8,500 JPY |
| System Plug-in | 50,800 JPY | 8,500 JPY |
| Variation Plug-in | 50,800 JPY | 8,500 JPY |
1年ライセンス
-
Basic Plug-in
358 USD -
Advanced Plug-in
358 USD -
System Plug-in
358 USD -
Variation Plug-in
358 USD
30日ライセンス
-
Basic Plug-in
59 USD -
Advanced Plug-in
59 USD -
System Plug-in
59 USD -
Variation Plug-in
59 USD
1年ライセンス
-
Basic Plug-in
358 EUR -
Advanced Plug-in
358 EUR -
System Plug-in
358 EUR -
Variation Plug-in
358 EUR
30日ライセンス
-
Basic Plug-in
59 EUR -
Advanced Plug-in
59 EUR -
System Plug-in
59 EUR -
Variation Plug-in
59 EUR
1年ライセンス
-
Basic Plug-in
50,800 JPY -
Advanced Plug-in
50,800 JPY -
System Plug-in
50,800 JPY -
Variation Plug-in
50,800 JPY
30日ライセンス
-
Basic Plug-in
8,500 JPY -
Advanced Plug-in
8,500 JPY -
System Plug-in
8,500 JPY -
Variation Plug-in
8,500 JPY
プラグインセット
| 1年ライセンス | |
|---|---|
| プラグインセット (2種セット) |
548 USD
|
| プラグインセット (3種セット) |
728 USD
|
| プラグインセット (4種セット) |
898 USD
|
- プラグインセットをご購入のお客様は、ご購入から一年間、データ解析サービス および オンライン・トレーニング をいつでも、何度でも4割引きでご利用いただけます。
-
プラグインを最大50% OFFでご利用いただける各種割引プログラムをご用意しています。
詳細はこちらをご覧ください。
割引プログラム一覧
| 1年ライセンス | |
|---|---|
| プラグインセット (2種セット) |
548 EUR
|
| プラグインセット (3種セット) |
728 EUR
|
| プラグインセット (4種セット) |
898 EUR
|
- プラグインセットをご購入のお客様は、ご購入から一年間、データ解析サービス および オンライン・トレーニング をいつでも、何度でも4割引きでご利用いただけます。
-
プラグインを最大50% OFFでご利用いただける各種割引プログラムをご用意しています。
詳細はこちらをご覧ください。
割引プログラム一覧
| 1年ライセンス | |
|---|---|
| プラグインセット (2種セット) |
76,800 JPY
|
| プラグインセット (3種セット) |
101,800 JPY
|
| プラグインセット (4種セット) |
125,800 JPY
|
- プラグインセットをご購入のお客様は、ご購入から一年間、データ解析サービス および オンライン・トレーニング をいつでも、何度でも4割引きでご利用いただけます。
-
プラグインを最大50% OFFでご利用いただける各種割引プログラムをご用意しています。
詳細はこちらをご覧ください。
割引プログラム一覧
-
プラグインセット(2種セット)
716 USD548 USD -
プラグインセット(3種セット)
1,074 USD728 USD -
プラグインセット(4種セット)
1,432 USD898 USD
プラグインセットをご購入のお客様は、ご購入から一年間、データ解析サービス および オンライン・トレーニング をいつでも、何度でも4割引きでご利用いただけます。
プラグインを最大50% OFFでご利用いただける各種割引プログラムをご用意しています。
詳細はこちらをご覧ください。
割引プログラム一覧
-
プラグインセット(2種セット)
716 EUR548 EUR -
プラグインセット(3種セット)
1,074 EUR728 EUR -
プラグインセット(4種セット)
1,432 EUR898 EUR
プラグインセットをご購入のお客様は、ご購入から一年間、データ解析サービス および オンライン・トレーニング をいつでも、何度でも4割引きでご利用いただけます。
プラグインを最大50% OFFでご利用いただける各種割引プログラムをご用意しています。
詳細はこちらをご覧ください。
割引プログラム一覧
-
プラグインセット(2種セット)
101,600 JPY76,800 JPY -
プラグインセット(3種セット)
152,400 JPY101,800 JPY -
プラグインセット(4種セット)
203,200 JPY125,800 JPY
プラグインセットをご購入のお客様は、ご購入から一年間、データ解析サービス および オンライン・トレーニング をいつでも、何度でも4割引きでご利用いただけます。
プラグインを最大50% OFFでご利用いただける各種割引プログラムをご用意しています。
詳細はこちらをご覧ください。
割引プログラム一覧
ご注文
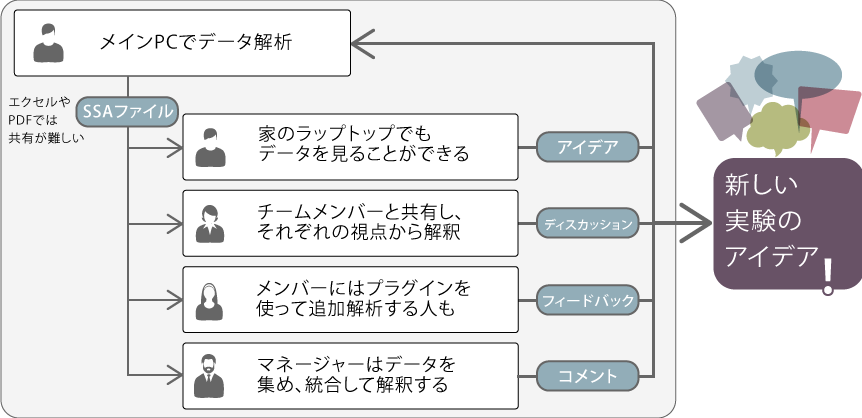
なぜプラグインシステムなのか?
プラグインを使って解析を実行するにはライセンスが必要ですが、Subio Platformに保存された解析結果を閲覧するのにライセンスは必要ありません。 データと解析結果を見るために、すべてのコンピューターにライセンスを購入する必要がないのです。この仕組みにより、従来のようにデータ解析を誰かに任せてしまうのではなく、解析を実行する人と解析結果を解釈する人が共同作業し、チーム全体で解析に取り込むことが可能となります。
解析を実行したら、すべての情報をたった一つのSSAファイルに出力することができます。 このSSAファイルを別のコンピューター上のSubio Platformで開けば、全く同じ状態を再現できます。 解析の続きを他の場所で行うことも、他の人に引き継いでもらうことも簡単にできます。 オミクスデータはとても複雑なので、たった一人の人がすべての意味を引き出すことはできません。したがって、誰でも再解析可能な状態でデータを共有することが、可能性を最大限に引き出すことになります。 Subio Platformのプラグインシステムは、このようなチームによる共同作業を可能にします。
 People and balloons are designed by Freepik.
People and balloons are designed by Freepik.