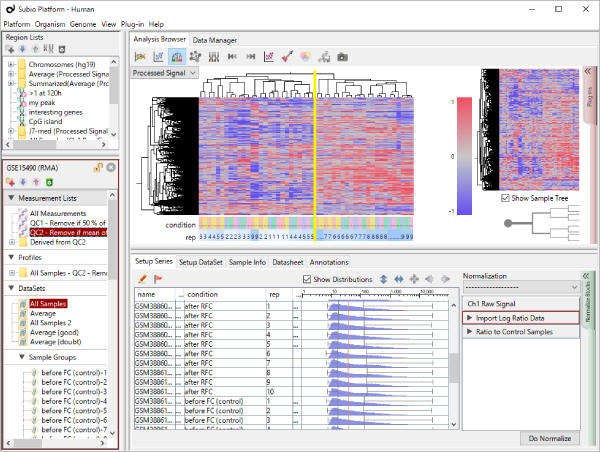
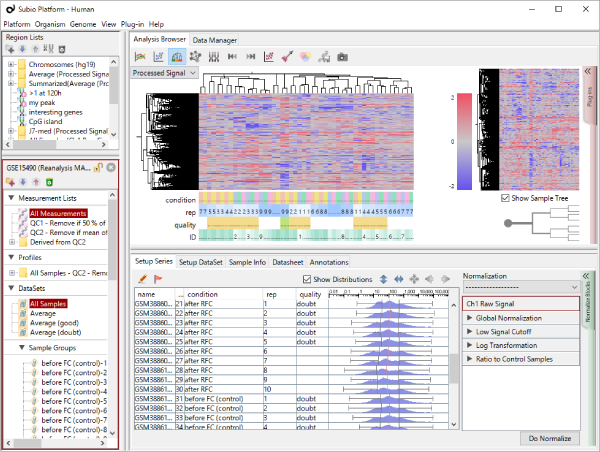
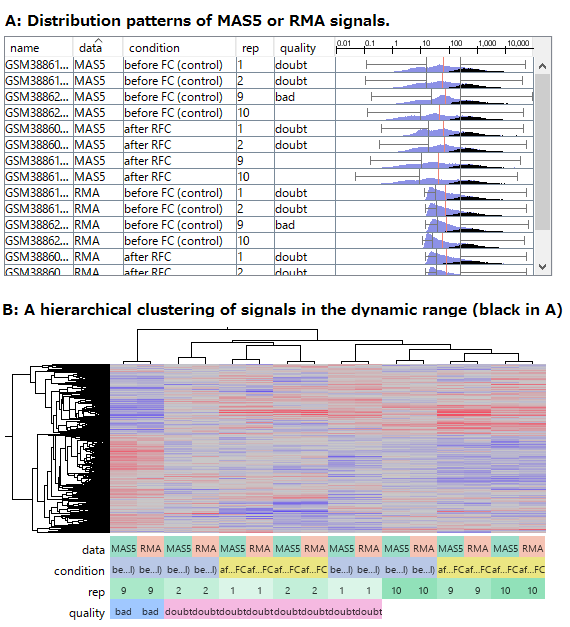
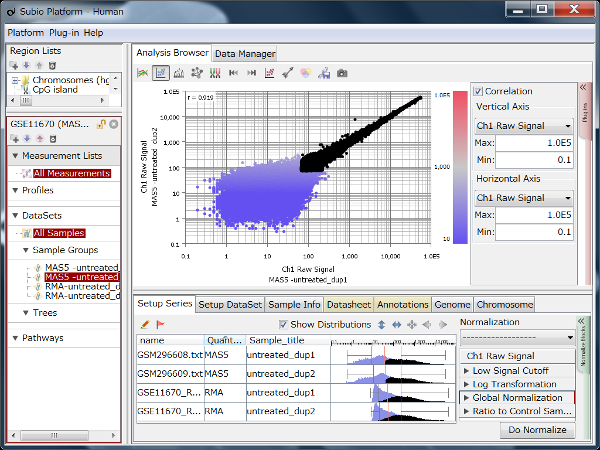
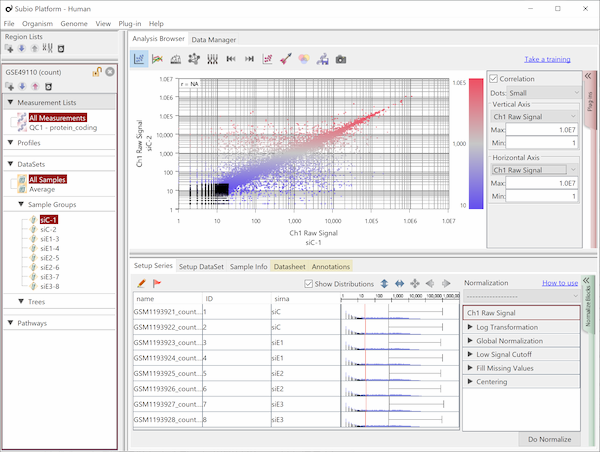
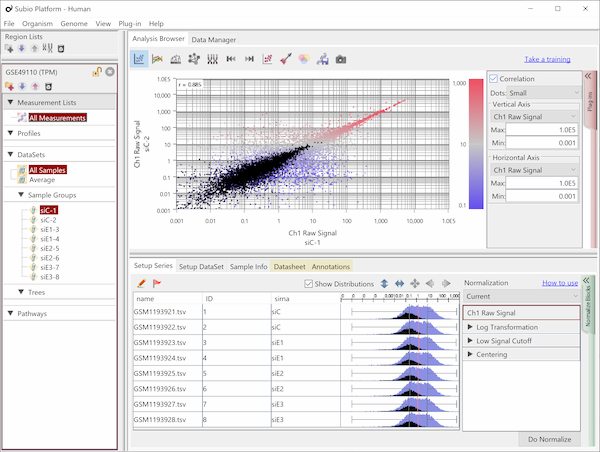
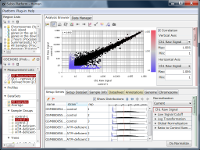
Let's say you are comparing technical replicate samples on a scatter plot.
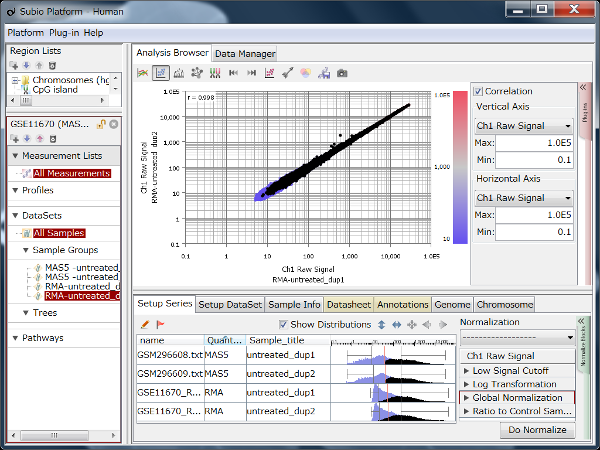
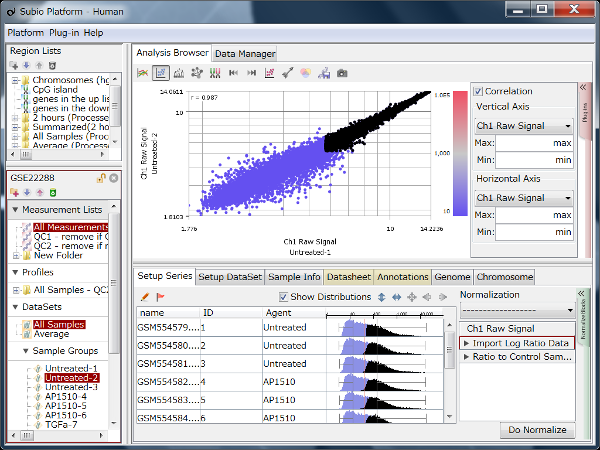
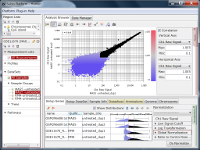
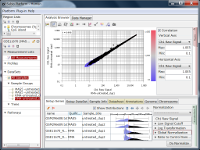
If you see charts like Fig4 and Fig5, which do you think is better quality?
Fig4 looks varying wider partially.
Correlation coefficient of Fig4 is 0.919, while that of Fig5 is 0.988.
I guess many think Fig5 is better.
But the two are generated from a same raw data and quantified by different algorithms, which are MAS5 (Fig4) and RMA (Fig5).
For more about the characteristics of the data, please see "MAS5 vs. RMA" for more about the characteristics of the data.
We focus on the reproducibility of microarray data here.
A high correlation coefficient indicates the high reproducibility.
But it is not always true when you talk about microarray data.
Why?
You cannot assume biologically that all genes are expressing in a sample.
And every measurement system has its own dynamic range.
You can rely on signals in the dynamic range (signal range), but cannot them out of the range (noise range).
A microarray data must have both signals and noise.
The noise is a mixture of not-expressing genes and too-low-to-detect by the system.
You cannot distinguish not-expressing and too-low-to-detect from noise.
So it is natural and reasonable there is an area where spots scatter widely at low signal range.
There is such a noise range in Fig4, but not in Fig5.
Actually, there is the noise, which is just hidden, from a view point of experimental biology.
Hiding noise is not welcome for analysts because it makes their works difficult.
If the noise range looks obvious, it is easy to set a boundary between noise and signal ranges, and extract genes with reliable signals (Fig4).
But it is not easy to do it in Fig5.
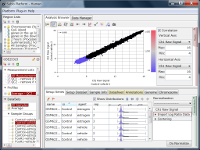
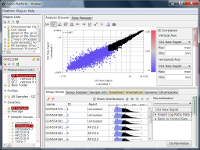
If you look closely at the histograms, there are peaks at their left ends.
And if you select genes in the signal range in Fig4, the shapes of them in histograms in Fig5 look similar.
So you can assume the left peaks are the noise range in Fig5, though it is hidden in the scatter plot.
A high correlation coefficient indicates the high reproducibility.
This is true for signal range, but not for noise range.
So a correlation coefficient calculated with all genes is not an issue.
This confusion resulted in algorithms hiding noise, or treating data as if all data are in the signal range.
You should clearly understand that the dynamic range is the signal range, excluding the noise range.
The noise is beneficial as it looks like noise.
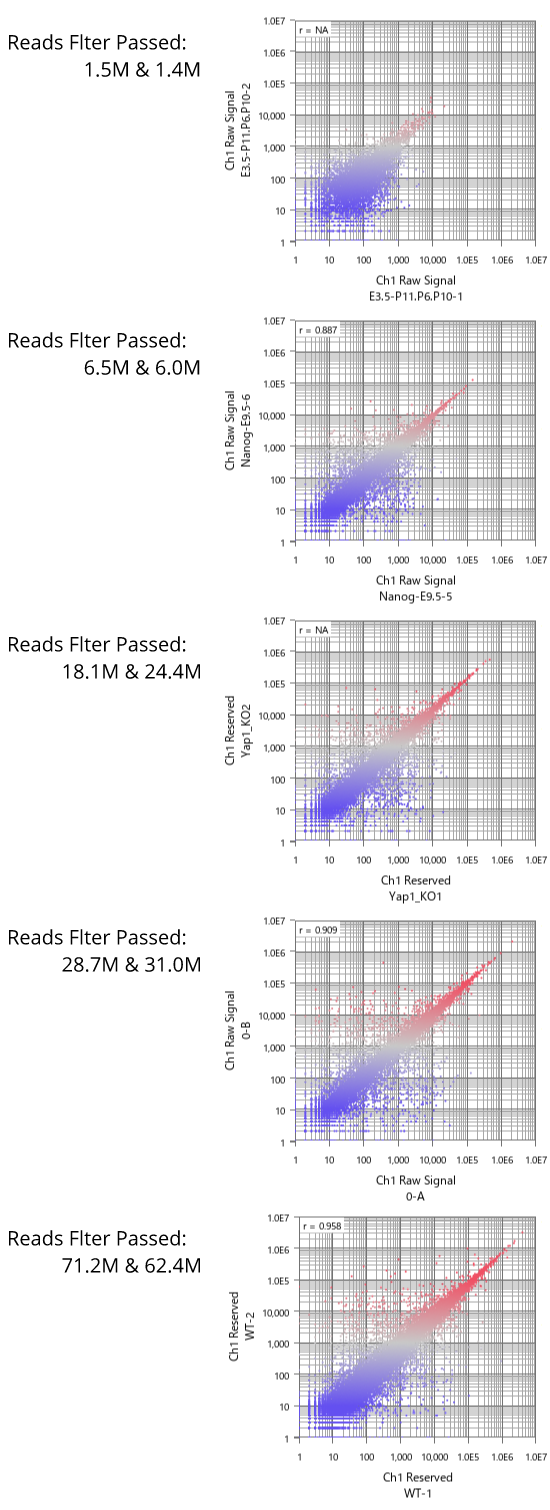
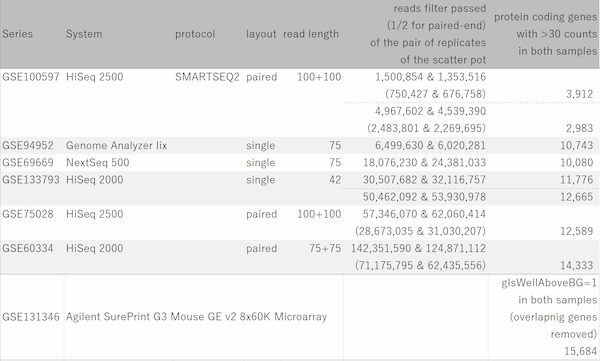
Now let's see dynamic ranges of microarray platforms.