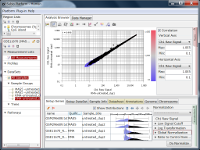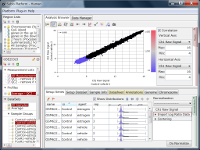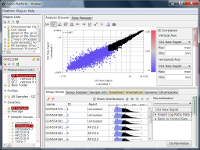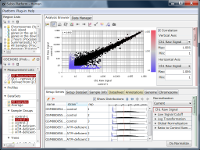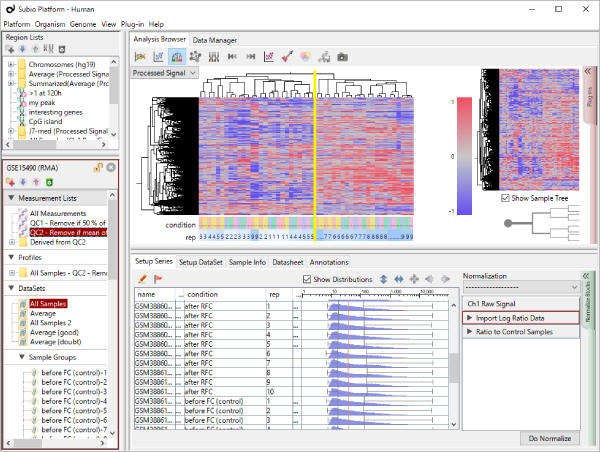
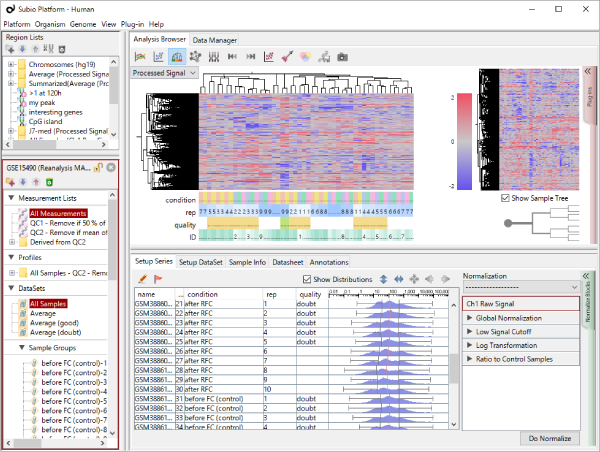
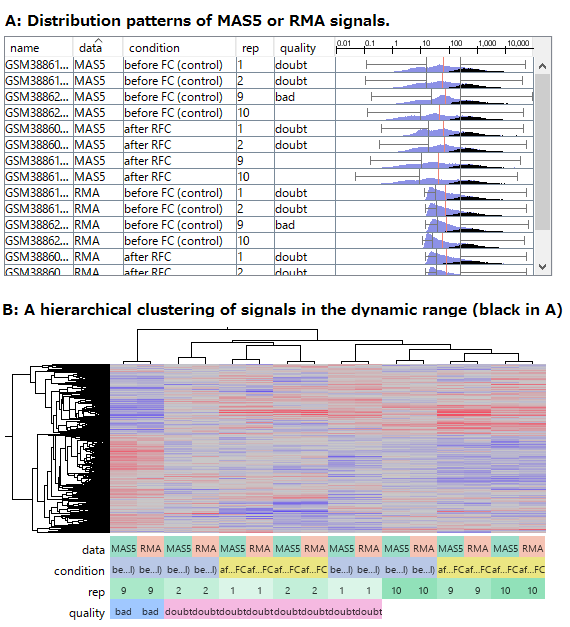
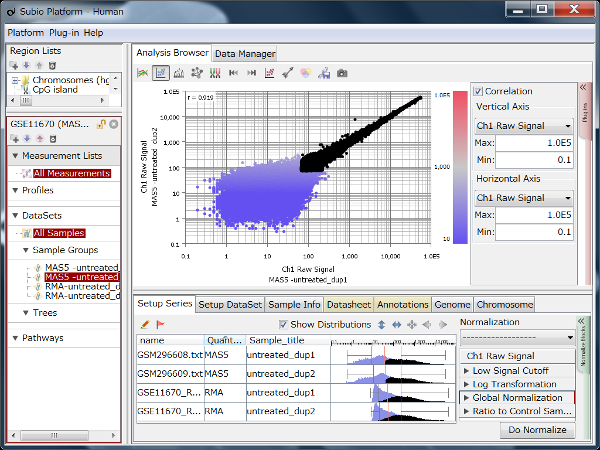
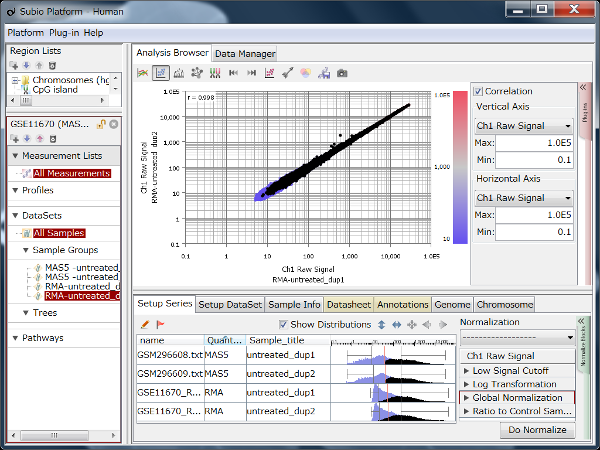
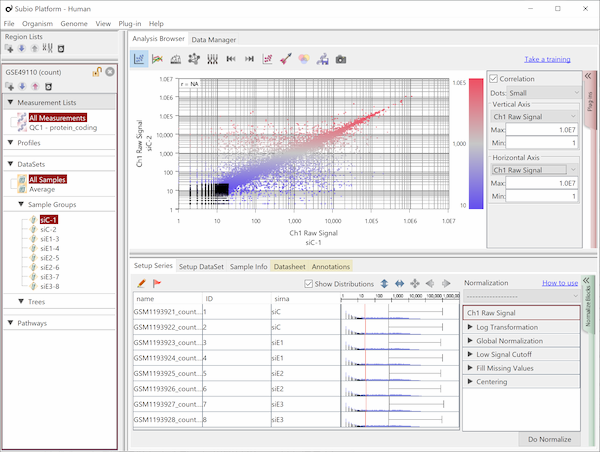
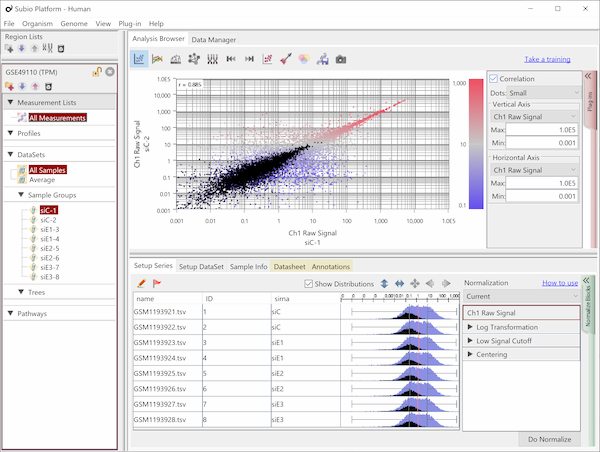
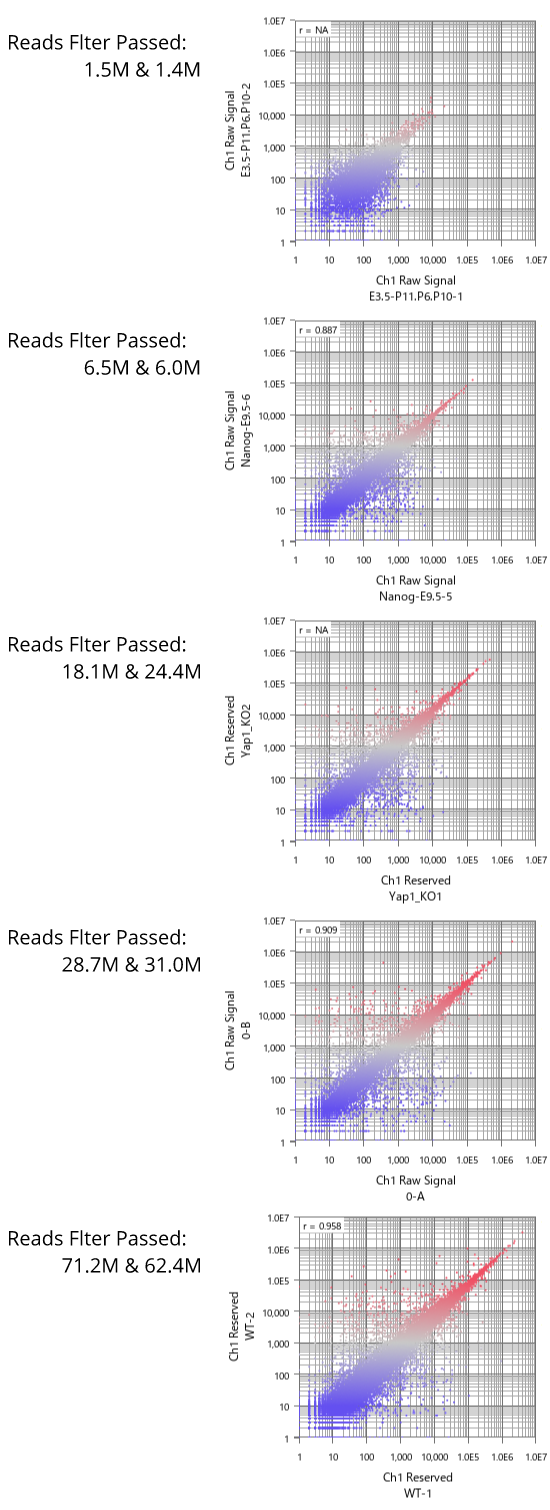
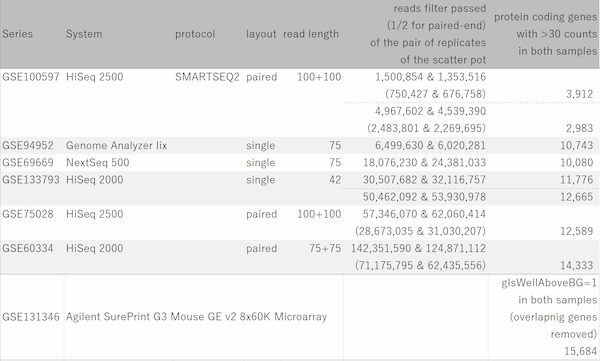
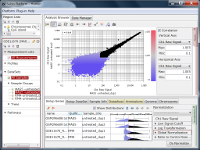
GSE11670もHG-U133 Plus2.0アレイを使った測定データです。 非常に質の高いテクニカルレプリケートの例です。 散布図の左下では大きなばらつきが見られますので、シグナル値が100より低い領域では測定値の信頼性が低いと言えます。 ヒストグラムを見ると、MAS5のシグナル値の分布は一山型に見えますが、実は本質的に二山形です。 シグナル領域とノイズ領域の重なる領域が広いので、ここにピークが見えているのです。 つまり、シグナル値が⒑から100の間の測定値は、信頼性は低いですが、完全にノイズではありません。 このデータは、SSAファイルをダウンロードしてお手元のSubio Platformで詳しくご覧いただけます。