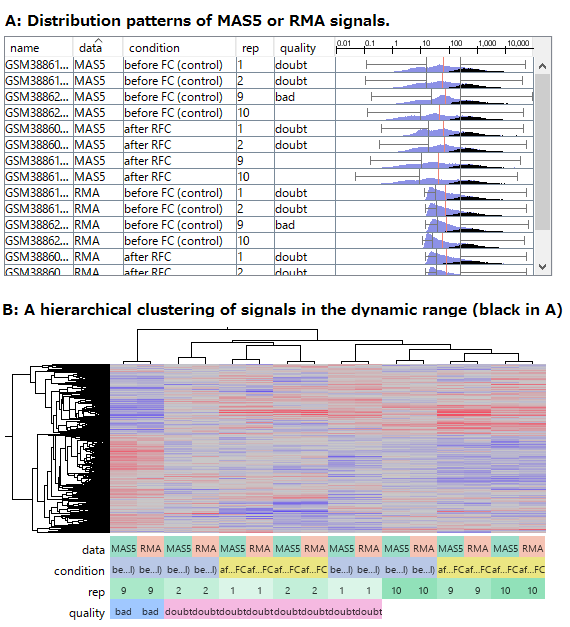
Fig3. RMAとMAS5のデータを、発現量の高い遺伝子だけで比較
- (A) RMA とMAS5 のシグナル値の分布の比較。黒はダイナミックレンジにあって、測定値が信頼できる発現量の高い遺伝子を表しています。
- (B) 発現量の高い遺伝子だけ(Aの黒)を使った階層型クラスタリングの結果。
このデータは、SSAファイルをダウンロードしてお手元のSubio Platformで詳しくご覧いただけます。
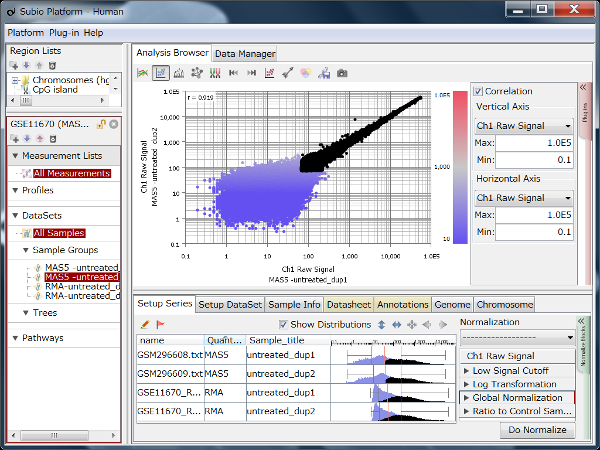
Fig4. Affymetrix HG-U133 Plus2.0 による測定データの例2(MAS5)
GSE11670もHG-U133 Plus2.0アレイを使った測定データです。 非常に質の高いテクニカルレプリケートの例です。 散布図の左下では大きなばらつきが見られますので、シグナル値が100より低い領域では測定値の信頼性が低いと言えます。 ヒストグラムを見ると、MAS5のシグナル値の分布は一山型に見えますが、実は本質的に二山形です。 シグナル領域とノイズ領域の重なる領域が広いので、ここにピークが見えているのです。 つまり、シグナル値が⒑から100の間の測定値は、信頼性は低いですが、完全にノイズではありません。 このデータは、SSAファイルをダウンロードしてお手元のSubio Platformで詳しくご覧いただけます。
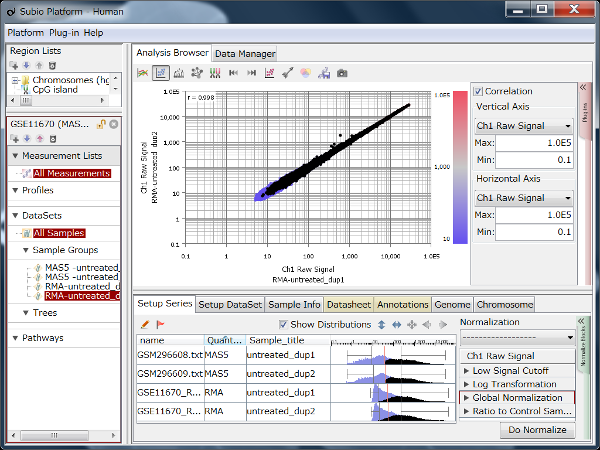
Fig5. Affymetrix HG-U133 Plus2.0 による測定データの例2(RMA)
Fig4と同じGSE11670の生データをRMAで再解析したものです。 散布図の左下に大きなばらつきが見えません。 しかし、だからと言ってこのデータにノイズがなく、すべての測定値がシグナルだというわけではありません。 ヒストグラムの3および4行目をご覧ください。 非常に高いピークが左端に見られます。 つまり、ノイズ領域にあった値を狭い領域に押し込めたにすぎないのです。 従って、シグナル値が10から100の領域は、MAS5の時よりノイズ成分が濃くなっているため信頼性が劣ります。
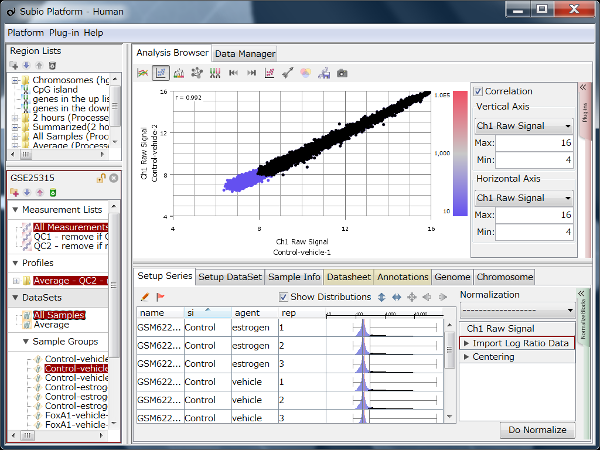
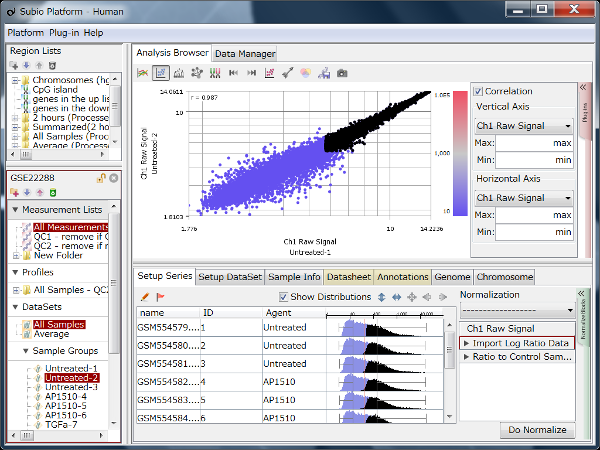
Fig7. Affymetrix Gene ST Arrayによる測定データの例
GSE22288は、Affymetrixが3' IVT Array(HG-U133 Plus2.0など)の後継として出したGene ST Arrayによる測定データです。 3' IVT Arrayでは、エクソンよりユニークな配列の多い3' UTRにプローブが設計されていたのに対し、Gene ST Arrayのプローブはエクソンに設計されています。 新しい技術が古い技術よりも良いとはかぎりません。 歴史的に見ても成熟した技術のほうが優れている例は少なくありません。 また、3' IVT GeneChipのデータはインターネットから大量に入手できるというメリットも考慮に値します。 このデータは、SSAファイルをダウンロードしてお手元のSubio Platformで詳しくご覧いただけます。
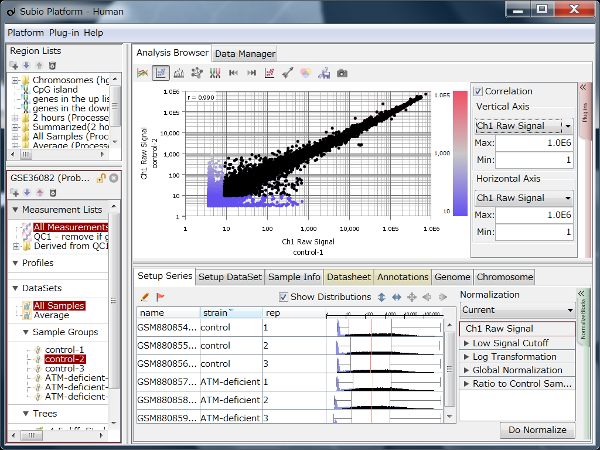
Fig8. Agilent Whole Genome 4x44kによる測定データの例
GSE36082は、マイクロアレイの歴史において大きな進歩を遂げたAgilent Whole Genome 4x44kによって測定された実験データです。 この後継であるSurePrintマイクロアレイも高い品質を維持しています。 ヒストグラムを見ると、左端に急峻なピークが見えます。 これはネガティブコントロースのプローブと、それと同等のシグナル値を示すプローブによって形成されるものです。 優れている理由は、ノイズ領域とシグナル領域の重複が少なく、境目がくっきりしていることです。 これにより、低くても発現している遺伝子と、発現していない遺伝子を区別して抽出できる可能性があるのです。 このデータは、SSAファイルをダウンロードしてお手元のSubio Platformで詳しくご覧いただけます。
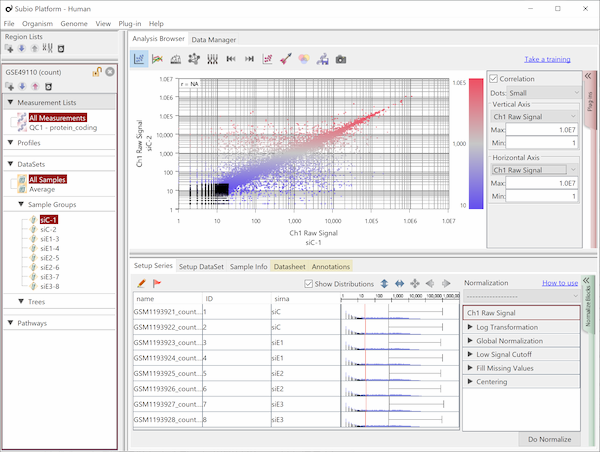
Fig9. Countの散布図
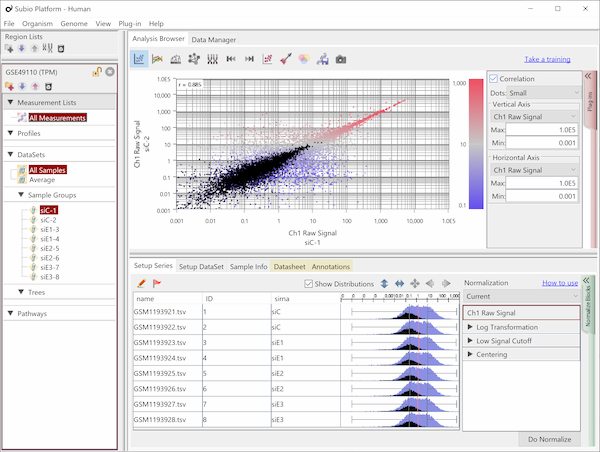
Fig10. TPMの散布図
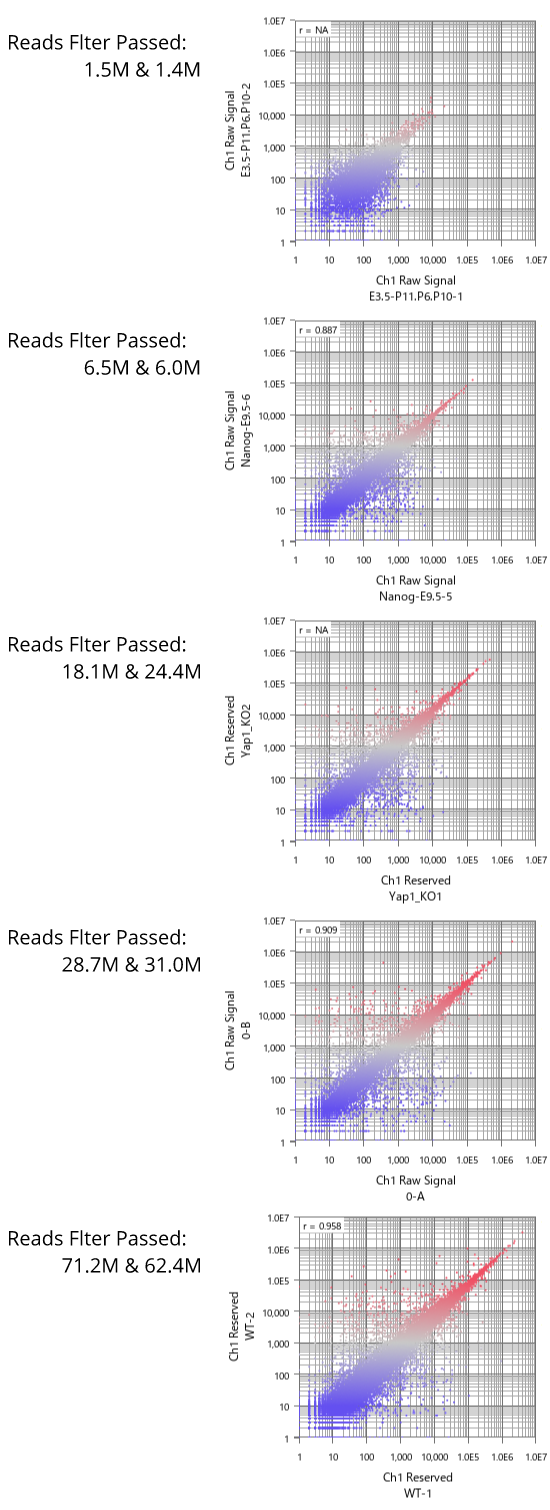
Fig11. RNA-Seqのリード数とダイナミックレンジの比較
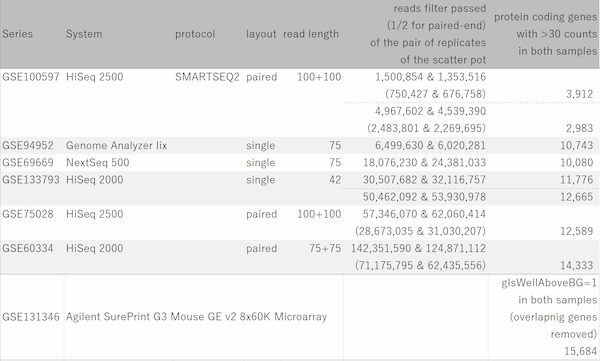
Fig12. RNA-Seqのリード数とダイナミックレンジの比較
- Fig1.
- Fig2.
- Fig3.
- Fig4.
- Fig5.
- Fig6.
- Fig7.
- Fig8.
- Fig9.
- Fig10.
- Fig11.
- Fig12.
オミクスデータにあるバイアス
たとえば、下表のように比較したい条件が5つあったとします。 それぞれの条件には繰返し実験が10サンプルずつあり、全部で50サンプルからなるデータセットがあります。
| グループ | サンプル数 |
|---|---|
| before_ R | 10 |
| after_ R | 10 |
| before_ FC | 10 |
| after_ FC | 10 |
| after_ RFC | 10 |
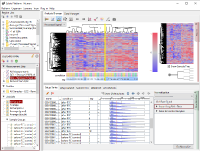
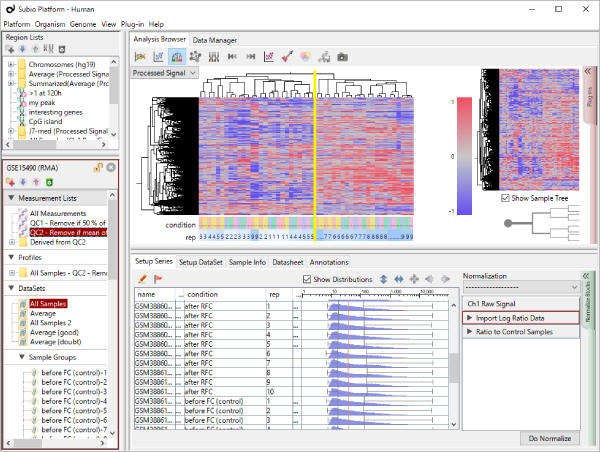
クラスタリングをしてみると、Fig1のような結果が得られました。 5つの条件を表すconditionの5色よりも、まず繰返し(rep)の前半(1~5)と後半(6~10)で大きく二つに分かれている(黄色の縦線)ことがわかります。 この結果を以て、sample 1~5と6~10の間で発現プロファイルに違いがあると結論付けるのは早計かもしれません。 生物学的に二つのサブグループに分かれるというよりは、なにか実験的な要因によるものではないかと考えるのが、実験生物学者としての直観ではないでしょうか。
もし実験的な要因によるものであれば、データに非線形のバイアスが含まれるので、グループ間の比較は単純に平均値を比較したりANOVA等の検定を適用したりするのは妥当ではありません。 二つの独立したデータセットとして解析してから、二つの結果をまとめて結論を出すなど、解析にはなんらかの工夫が必要です。
MAS5とRMA
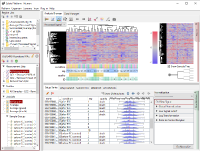
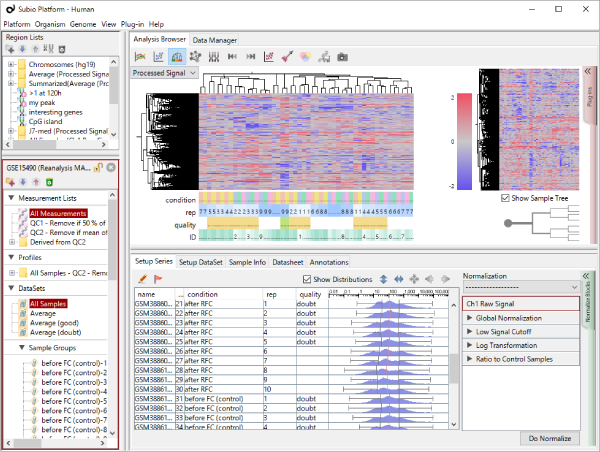
Fig1のヒストグラムは、シグナル値の分布を表しています。 ただしRMAという手法により数値化されているデータなので、強力な正規化が施されたシグナル値と言ったほうが適切です。 より生データに近い値を見たければ、MAS5という手法で数値化をやり直してみるといいです(Fig2)。
Fig2とFig1はまったく同じ生データから得られたシグナル値ですが、数値化手法の違いによってデータがまるで違って見えます。 ヒストグラムよく見ると、rep 1~5とrep 6~10では25th percentileから75th percentileの範囲が違うことがわかります。 より生データに近いMAS5 (Fig2)では、rep 6~10ではダイナミックレンジが広くて(100より上の領域)、rep 1~5では狭く(200より上の領域)なっています。 ちなみに、ダイナミックレンジとはすべての値の分布域ではなく、きちんと測定されているシグナル領域の分布域のことです。 シグナル値が100付近の遺伝子は、後半のサンプルではきちんと測定されているのに対して、前半のサンプルでは測定値は疑わしいということです。 この差がRNAの品質によるものなのか、アレイの染色や洗いなど実験過程によるものなのかはわかりませんが、いずれにせよダイナミックレンジの差が発現プロファイルに影響を与えていて、Fig1のクラスタリングの原因になっていると考えられます。
このケースのように、生データの分布の形が違う場合でも強制的に形を揃えるためにQuantile Normalizationなど強力な正規化手法が開発されました。 RMA (Fig1)はQuantile Normalizationを含んでいるので、MAS5 (Fig2)よりヒストグラムの形がそろっています。 しかし、どのような強力な正規化手法を使っても、生データの性質の違いをなかったことにはできないということを知るべきです。

- (A) RMA とMAS5 のシグナル値の分布の比較。黒はダイナミックレンジにあって、測定値が信頼できる発現量の高い遺伝子を表しています。
- (B) 発現量の高い遺伝子だけ(Aの黒)を使った階層型クラスタリングの結果。
このデータは、SSAファイルをダウンロードしてお手元のSubio Platformで詳しくご覧いただけます。
ところで、RMA (Fig1) と MAS5 (Fig2) のデータをさらによく見てみると、ヒストグラムの形が一見まったく違って見えるにも関わらず、シグナル値が十分に高く測定値が信頼できる領域(ダイナミックレンジ)にある値だけを見ると、非常によく似ていることがわかります (Fig3 Aの黒)。 この領域の遺伝子だけを使えば、RMAもMAS5も解析結果にほとんど違いはありません (Fig3 B)。 二つのアルゴリズムで違ってくるのは、ノイズ領域の値 (Fig3 Aの青) に過ぎないことがわかります。
これらのデータについては、右のFig1〜3にSSAファイルのリンクがありますので、ダウンロードしてお手元のSubio Platformで詳しくご覧ください。
RMAの欠点
MAS5とRMAで違いが現れるのは、シグナル値の低い領域です。 この領域において、MAS5とRMAのどちらが優れているかについてはここで議論しません。 しかし解析結果を解釈する立場から言うと、RMAには看過できない欠点が二つあります。
まず、上述のとおり非線形のバイアスは見えにくくなるだけで、本質的には残ったままです。 つまりそのデータの解釈には、経験と注意深さがより一層求められます。 熟練した解析者であれば、クラスタリングやPCAの結果を見て非線形バイアスの存在を察知して、それを裏付けるために実験者、日付、場所、その他なんらかの実験条件が関与していないかを調べることができます。 しかしほとんどの解析者にとって、バイアスを隠すことは拙速に誤った結論へと導くことになります。
次に、測定値が信頼できるシグナルの領域と、測定値が信頼できないノイズの領域の区別をつけるのが非常に難しいということです。 マイクロアレイにはあらかじめすべての遺伝子に対するプローブが設計されていますが、すべての遺伝子が発現しているは生物学的には考えられません。 発現していない遺伝子や、発現が低すぎて信頼できる測定値が得られない領域が必ずあるはずなのに、それが見えにくくなります。 ノイズを隠すこともまた、誤った結論に導くことになります。 詳しくは、マイクロアレイのダイナミックレンジについての解説をご覧ください。
さらにRMAには、実験計画上の適用範囲を狭める制限が二つあります。 まず「全てのサンプルにおける発現プロファイルが殆ど変わらない」という前提が成り立たないとき、そして前向き研究などで新しいサンプルがどんどん追加されるようなときにはRMAは不適切です。 前者のケースとは、たとえば異なる種類の細胞を比べるとき、異なる細胞組成の組織サンプルを比べるとき、異なる発生段階を比べるとき、異なる進行度の病変サンプを比べるときなど、比較的多くの研究において当てはまることなので、決して特殊なケースとは言えません。
以上より、解析結果の解釈および実験計画の観点からはMAS5のほうがRMAよりも優れているのですが、Affymetrix(現Thermo Fisher Scientific)の新しいGeneChipではMAS5による解析ができなくなってしまいました。 バイオロジ―不在のバイオインフォマティクスがもたらした退化だと思います。
大事なのは、質の高い生データ
このようなことを書いているのは、特定のアルゴリズムや技術について悪く言うためではありません。 不合理な伝説が蔓延ることで生命科学の実施的な進歩が阻まれる状況になっている一例を示しているのです。 データをきれいに見せる手法はいろいろありますが、バイアスを隠しているだけで本当に取り除くことはできません。 実験生物学の視点から言うと、そのようなアルゴリズムに頼らないで済むよう「質の高い生データを取得するためにはどうするか」をよく考えて実験すること、そしてデータをありのままに見せてくれるアルゴリズムを使うことに尽きます。
私たちの提案するソリューション、Subio Platform

オミクスデータをどう解析すればいいのか、知っている人は一人もいません。 解析の専門家などいないのです。 おそらく決定的なアイデアを持ち込んでくるのは、統計学者やバイオインフォマティシャンではなく、実験と現象に精通している実験生物学者でしょう。 彼らが積極的に加わって、すべての参加者が等しくアマチュアとしてアイデアを出し合って検討しなければ、オミクスを活用した新しい生命科学はこれ以上先に進むことができないというところに来ています。
私たちがこの問題にどのように向き合い、なぜSubio Platformを提案しているかご覧ください。
よろしければ、こちらも合わせてお読みください。