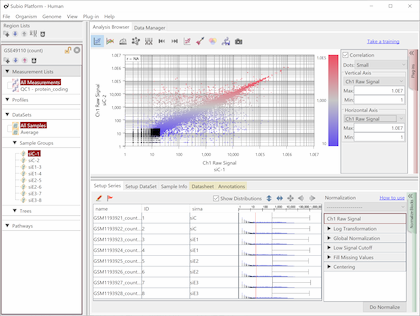
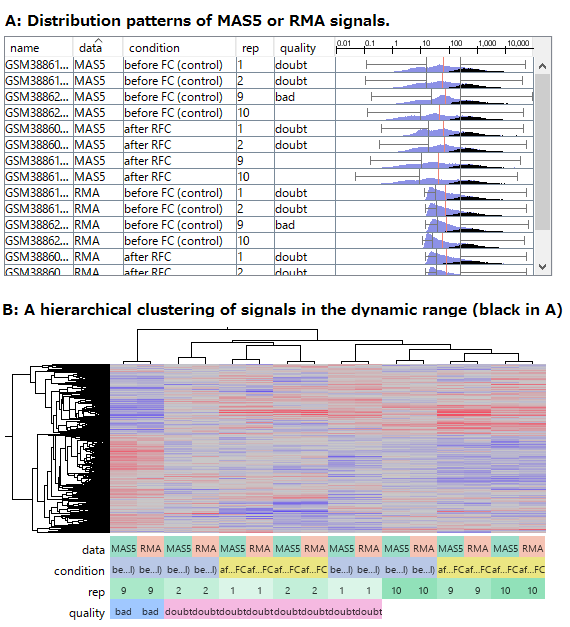
Fig3. RMAとMAS5のデータを、発現量の高い遺伝子だけで比較
- (A) RMA とMAS5 のシグナル値の分布の比較。黒はダイナミックレンジにあって、測定値が信頼できる発現量の高い遺伝子を表しています。
- (B) 発現量の高い遺伝子だけ(Aの黒)を使った階層型クラスタリングの結果。
このデータは、SSAファイルをダウンロードしてお手元のSubio Platformで詳しくご覧いただけます。
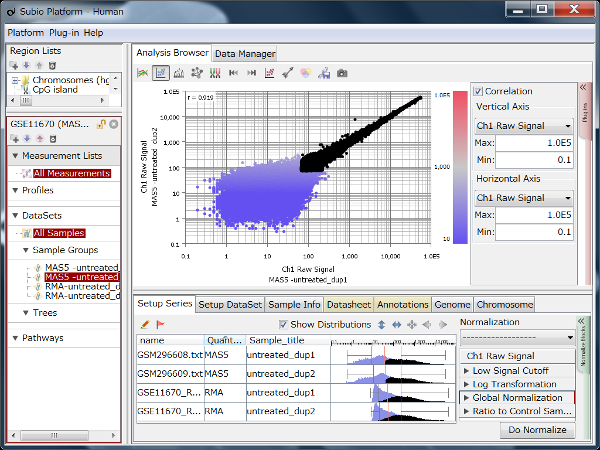
Fig4. Affymetrix HG-U133 Plus2.0 による測定データの例2(MAS5)
GSE11670もHG-U133 Plus2.0アレイを使った測定データです。 非常に質の高いテクニカルレプリケートの例です。 散布図の左下では大きなばらつきが見られますので、シグナル値が100より低い領域では測定値の信頼性が低いと言えます。 ヒストグラムを見ると、MAS5のシグナル値の分布は一山型に見えますが、実は本質的に二山形です。 シグナル領域とノイズ領域の重なる領域が広いので、ここにピークが見えているのです。 つまり、シグナル値が⒑から100の間の測定値は、信頼性は低いですが、完全にノイズではありません。 このデータは、SSAファイルをダウンロードしてお手元のSubio Platformで詳しくご覧いただけます。
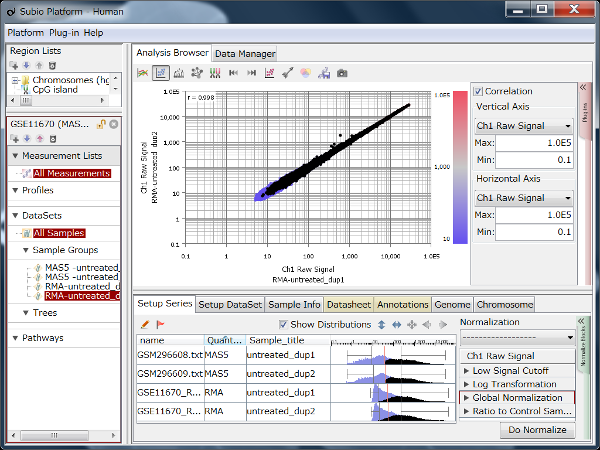
Fig5. Affymetrix HG-U133 Plus2.0 による測定データの例2(RMA)
Fig4と同じGSE11670の生データをRMAで再解析したものです。 散布図の左下に大きなばらつきが見えません。 しかし、だからと言ってこのデータにノイズがなく、すべての測定値がシグナルだというわけではありません。 ヒストグラムの3および4行目をご覧ください。 非常に高いピークが左端に見られます。 つまり、ノイズ領域にあった値を狭い領域に押し込めたにすぎないのです。 従って、シグナル値が10から100の領域は、MAS5の時よりノイズ成分が濃くなっているため信頼性が劣ります。
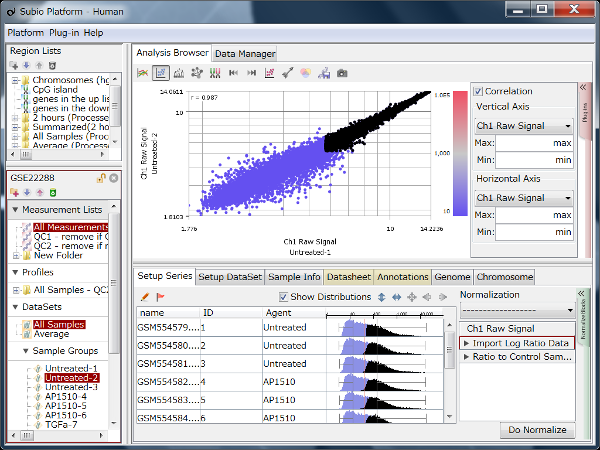
Fig7. Affymetrix Gene ST Arrayによる測定データの例
GSE22288は、Affymetrixが3' IVT Array(HG-U133 Plus2.0など)の後継として出したGene ST Arrayによる測定データです。 3' IVT Arrayでは、エクソンよりユニークな配列の多い3' UTRにプローブが設計されていたのに対し、Gene ST Arrayのプローブはエクソンに設計されています。 新しい技術が古い技術よりも良いとはかぎりません。 歴史的に見ても成熟した技術のほうが優れている例は少なくありません。 また、3' IVT GeneChipのデータはインターネットから大量に入手できるというメリットも考慮に値します。 このデータは、SSAファイルをダウンロードしてお手元のSubio Platformで詳しくご覧いただけます。
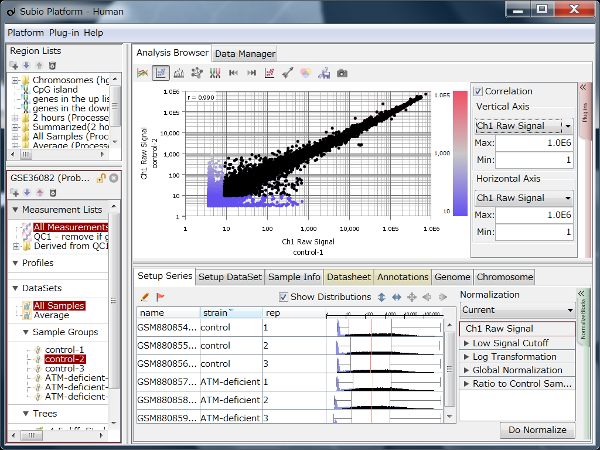
Fig8. Agilent Whole Genome 4x44kによる測定データの例
GSE36082は、マイクロアレイの歴史において大きな進歩を遂げたAgilent Whole Genome 4x44kによって測定された実験データです。 この後継であるSurePrintマイクロアレイも高い品質を維持しています。 ヒストグラムを見ると、左端に急峻なピークが見えます。 これはネガティブコントロースのプローブと、それと同等のシグナル値を示すプローブによって形成されるものです。 優れている理由は、ノイズ領域とシグナル領域の重複が少なく、境目がくっきりしていることです。 これにより、低くても発現している遺伝子と、発現していない遺伝子を区別して抽出できる可能性があるのです。 このデータは、SSAファイルをダウンロードしてお手元のSubio Platformで詳しくご覧いただけます。
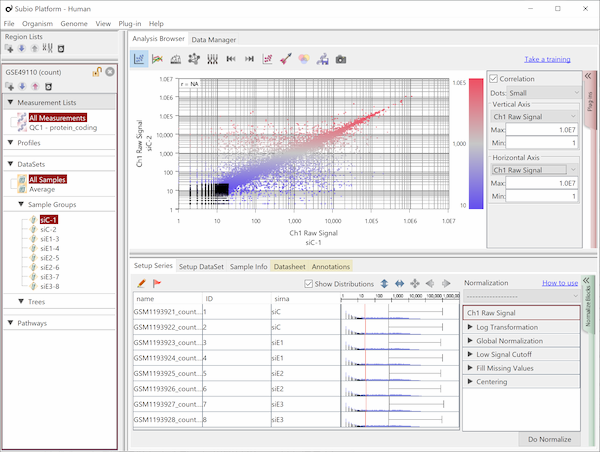
Fig9. Countの散布図
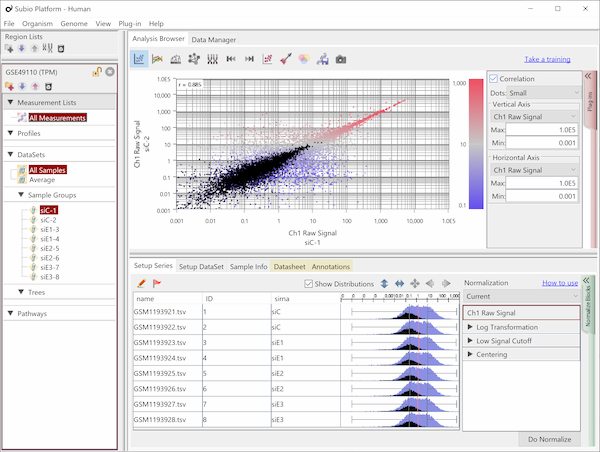
Fig10. TPMの散布図
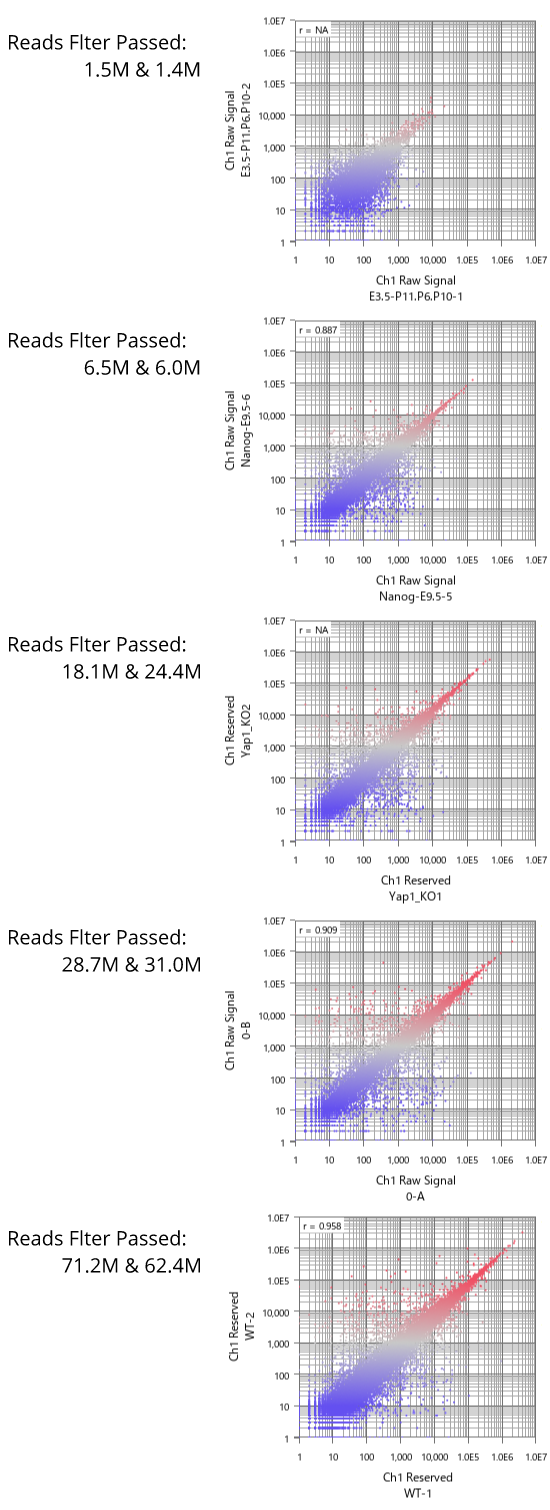
Fig11. RNA-Seqのリード数とダイナミックレンジの比較
Fig12. RNA-Seqのリード数とダイナミックレンジの比較
- Fig1.
- Fig2.
- Fig3.
- Fig4.
- Fig5.
- Fig6.
- Fig7.
- Fig8.
- Fig9.
- Fig10.
- Fig11.
- Fig12.
RNA-Seqのダイナミックレンジ
RNA-Seqによる発現量のデータには2種類あります。 まずはリードカウント(またはCount)であり、もう一つはTPM・FPKM・RPKMです。
TPM・FPKM・RPKMは、Countに対して、100万リードあたりの正規化と、1kbあたりの正規化を適用した値です。 RNA-Seqデータ解析の解説の多くが、リードカウントではなく、TPM・FPKM・RPKMの使用を推奨していることから、論文や学会発表ではこれらを用いた解析結果を目にすることが多いです。
しかし、RNA-Seqのデータを正しく解析するには、この違いを理解して適切に選ぶ必要があります。
Countについて
Fig9はHiSeq2000によるRNA-Seqのデータで、繰り返しサンプルのCountを散布図で表したものです。 Countの値が20より高い領域で測定値が対角線上に収束しており、再現性が高いことを示唆しています。 しかし、Countの値が20より低い領域ではばらつきが大きくなっています。 測定値が信頼できないノイズ領域だということです(黒点)。 ダイナミックレンジとは、すべての値の分布域のことではなく、ノイズ領域を除いたシグナル領域(色のついた点)のことです。
ここでは便宜的に20という閾値を使って説明していますが、実際にはノイズ領域とシグナル領域の境は明確なものではなく、グラデーションです。 このデータでいうと、10から100の間で、値が低くなるほど測定値の信頼性が落ちていくと言えるでしょう。
TPM・FPKM・RPKMについて
1kbあたりの正規化について
Fig10はFig9と同じデータですが、値はTPMです。 黒点はFig9のノイズ領域にあった遺伝子群と同じです。 対角線方向に広がっていて、シグナル領域の遺伝子群と区別することができないことにお気づきでしょう。 しかし、ノイズ領域を持たない測定システムは存在しません。
リードカウントは遺伝子の大きさを反映して、大きな遺伝子ではCountが多くなり、逆に小さな遺伝子ではCountが小さくなることが予想されます。 そこで、エクソン長1kbpあたりで均すのが良いと言われています。 この主張は一見妥当に見えるでしょうが、よく考えてみてください。
長さで均すということは、さきほど見たCountの点が、大きい遺伝子は左下方向に、小さな遺伝子は右上方向に、対角線に沿って移動することになります。 これが、ノイズ領域が見えなくなる効果を生んでいます。 しかも、エクソン長の平均は1kbよりも長いので、多くの遺伝子は左下方向に移動し、値の範囲も大きく広がります。 しかし、これは1kbあたりの正規化をした副作用に過ぎません。 この副作用を以て「ダイナミックレンジが広い」や「低発現領域に強い」と主張する論文が多いので注意が必要です。
つまり、ダイナミックレンジを検討するなら、TPM・FPKM・RPKMではなく、Countを見なければいけません。
ところで、「1kbあたりで均さないと発現解析に適さない」という主張は本当でしょうか。 たしかに、同一サンプルにおける遺伝子間の発現量の多寡を知りたいのであれば、1kbで正規化する必要があります。 しかし、同一遺伝子のサンプル間における発現量の変化が解析対象であれば、長さの正規化は必要はありませんね。 また、1kbあたりで均すことによって、データ解析がノイズの影響を受けやすくなります。
RNA-Seqのリード数とダイナミックレンジ
RNA-Seqのダイナミックレンジは、原理的にリード数に依存します。 リード数が少なければ、ほとんどのリードは発現が極めて高い少数の遺伝子によって占められます。 低発現の遺伝子の発現量を測定するには、膨大な数のリードが必要だと予想できるでしょう。 それでは公共データベースを使って、目的とするダイナミックレンジにどれくらいのリード数が必要かを見ていきましょう。
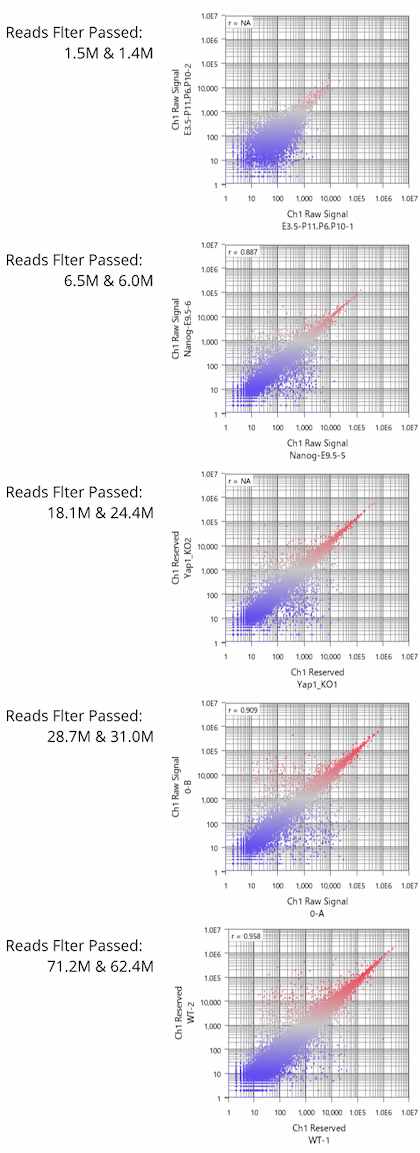
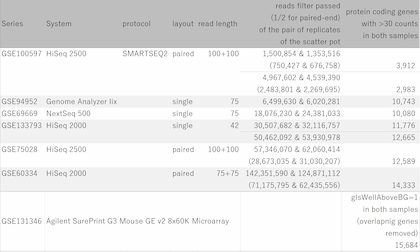
Fig11は、GEOからリード数の異なるデータセットをいくつか選び出して、繰り返しサンプルにおけるCount値を散布図で視覚化したものです。 予想されたとおり、リード数が増えるほどダイナミックレンジが広がっていく様子が分かります。 そして、もう少し詳しくまとめたのが下表です。
ダイナミックレンジが広いほど、より多くの遺伝子がシグナル領域に入ってきます。 フィルターのかけ方によって数は変わりますので、表中に示した遺伝子数は、シグナル領域にある遺伝子数のおおまかな目安だと思って下さい。
リード数が1000万くらいあると、発現の高いほうから1万個くらいの遺伝子が解析対象に入ってきます。 しかし、そこからダイナミックレンジを伸ばそうとすると、必要なリード数は飛躍的に増加することが分かります。
リード数だけでなく、インプットRNAの量も、ダイナミックレンジにきわめて大きな影響を持っています。 超微量インプットのRNA-Seqやsingle cell RNA-Seqでは、ダイナミックレンジが極めて狭くなりがちです。 技術の進歩によって改善される傾向にありますが、インプット量の多いときよりも測定データの品質にはばらつきが出やすく、安定したデータ得るためには細心の注意が必要です。
また、複数のデータセットを見ていると、ほとんどのデータセットにおいて最大リード数と最小リード数には2倍くらいの差があることが分かります。 データ解析は、ダイナミックレンジの狭いサンプルに合わせて調整する必要がありますので、リード数を見積もる際はシーケンサーのカタログ上の理論値を当てにするのは危険です。 理論値の半分くらいを想定して実験計画を策定するのが現実的でしょう。
3' 末端付近のみをシーケンスにかけることで、通常のRNA-Seqと比べてダイナミックレンジが改善するということは、原理的にありえません。 実際に測定を行ったところ、再現性に違いは見られないものの短い転写物がより多く検出されたという報告があります。 つまり 3' RNA-Seq は、短い遺伝子に興味があるのであれば良い選択肢となりますが、一部の広告が謳っている「リード数を抑えることができる」効果はありません。
最後に、UMIについてお話します。 UMIは、PCRによる不均一な増幅を補正するための仕組みです。 しかし、同一のUMIをまとめる(deduplication)ということは、リード数減少という副作用をもたらします。 つまりUMIは、ダイナミックレンジを犠牲にするトレードオフとして考えるべきです。 もしリード数が必要数の10倍以上あるのであれば、UMIによるプラスの効果を享受できるでしょう。 しかし、single-cell RNA-Seqや超微量インプットからのRNA-Seqではそもそもリード数が足りていません。 このような状況においてデータ解析パイプラインにUMI処理を組み込むことは、データの品質を決定的に損なう可能性があります。
よろしければ、こちらも合わせてお読みください。